pbmc_cell_frequency | Removed 550 specimens because missing in meta data
plasma_ab_titer | Removed 6931 specimens because missing in meta data
plasma_cytokine_concentration_by_olink | Removed 495 specimens because missing in meta data
pbmc_cell_frequency | Removed 56 features because not in feature subset
plasma_ab_titer | Removed 48 features because not in feature subset
plasma_cytokine_concentration_by_olink | Removed 234 features because not in feature subset
t_cell_polarization | Removed 3 features because not in feature subset
plasma_cytokine_concentration_by_olink | Removed 300 features because qc warning
plasma_ab_titer | Removed 10540 measurements because wrong unit used
plasma_cytokine_concentration_by_olink | Removed 2400 measurements because wrong unit used
plasma_cytokine_concentration_by_legendplex | Removed 8 because specimen is outlier
plasma_cytokine_concentration_by_olink | Removed specimen 750, 760, 824, 833, 894, 903 because fraction of measurements below LOQ > 50%
plasma_ab_titer | Removed specimen 674, 675, 676 because fraction of measurements below LOD > 50%
converting counts to integer mode
Found 4 batches
Using null model in ComBat-seq.
Adjusting for 0 covariate(s) or covariate level(s)
Estimating dispersions
Fitting the GLM model
Shrinkage off - using GLM estimates for parameters
Adjusting the data
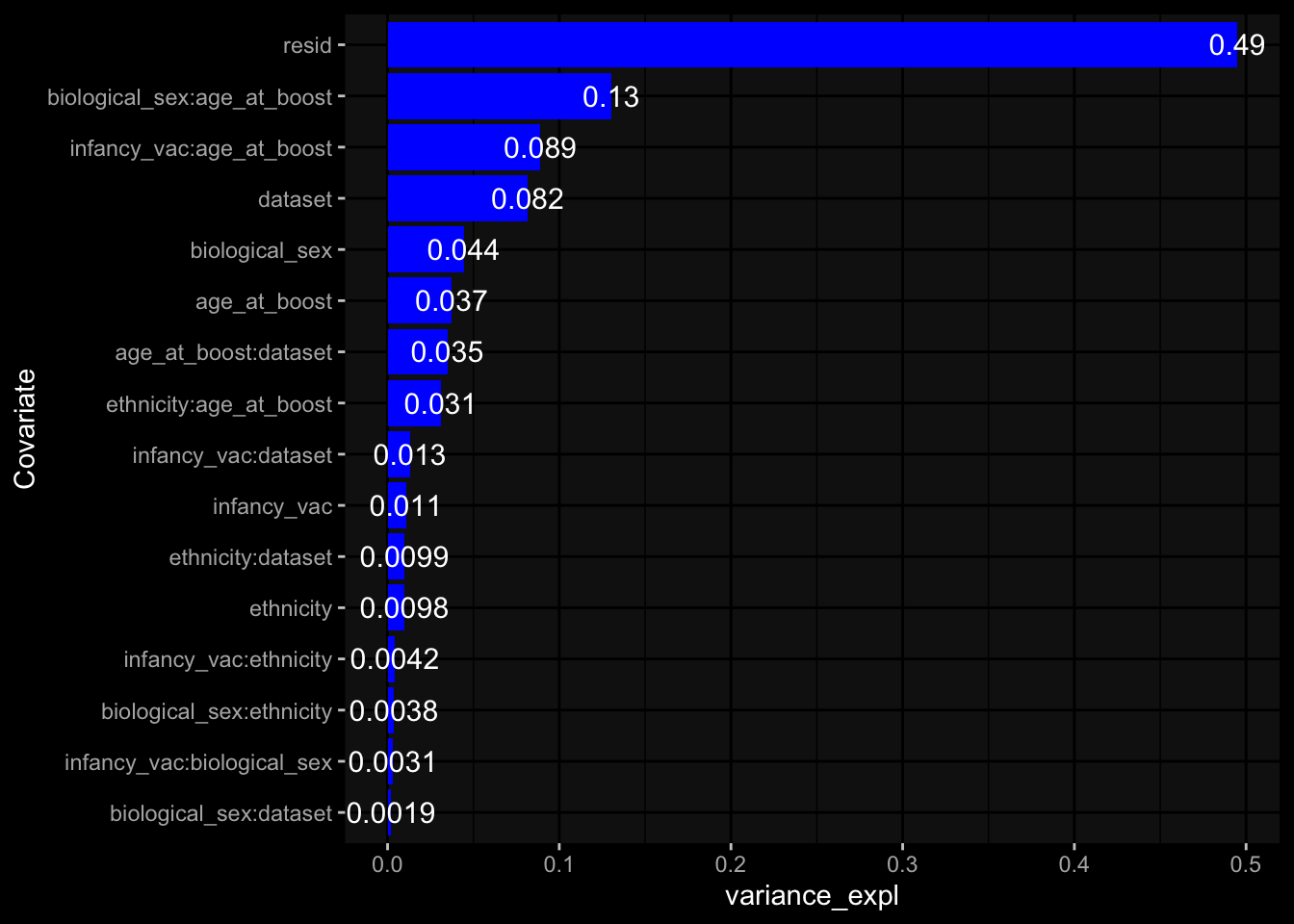
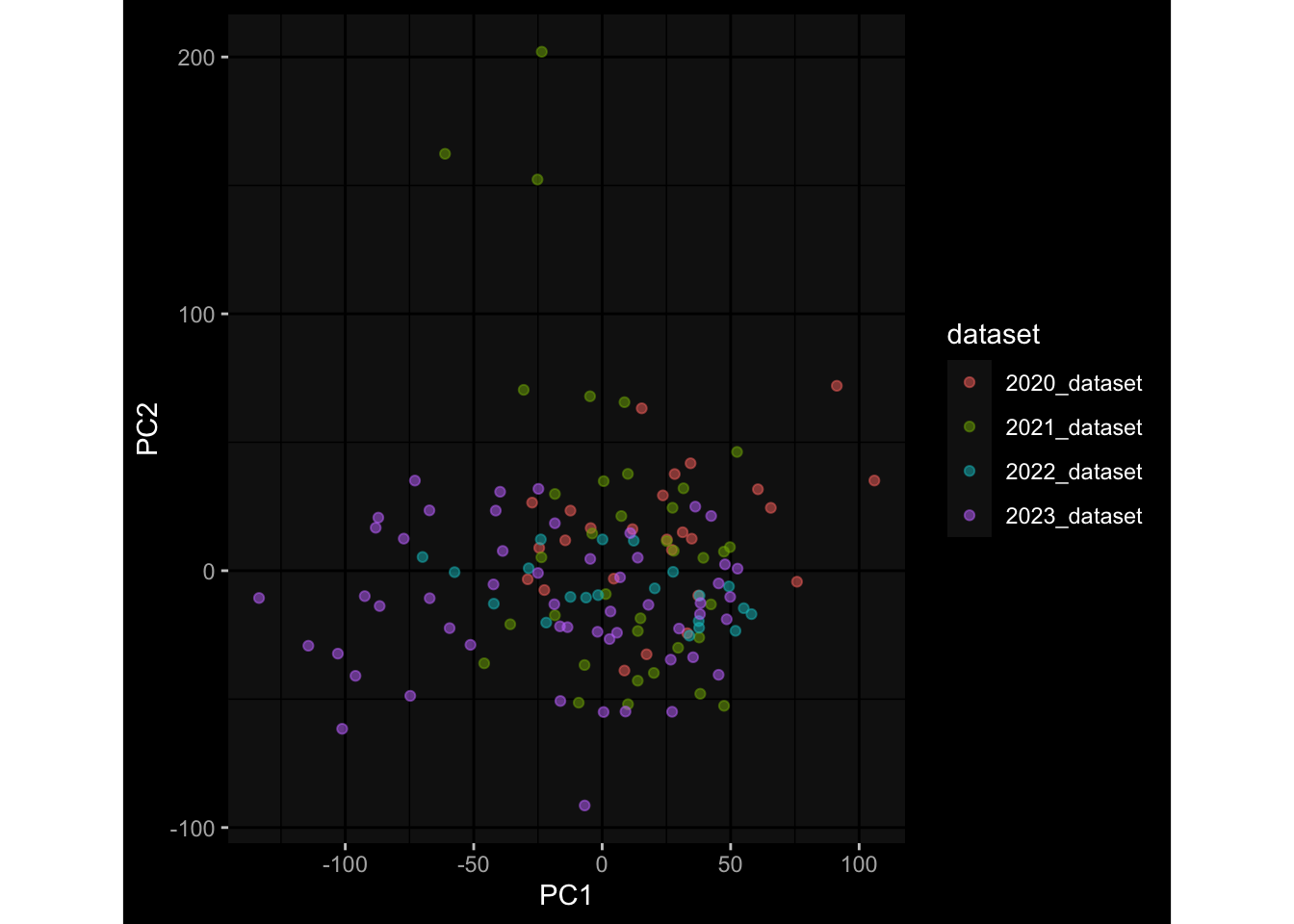
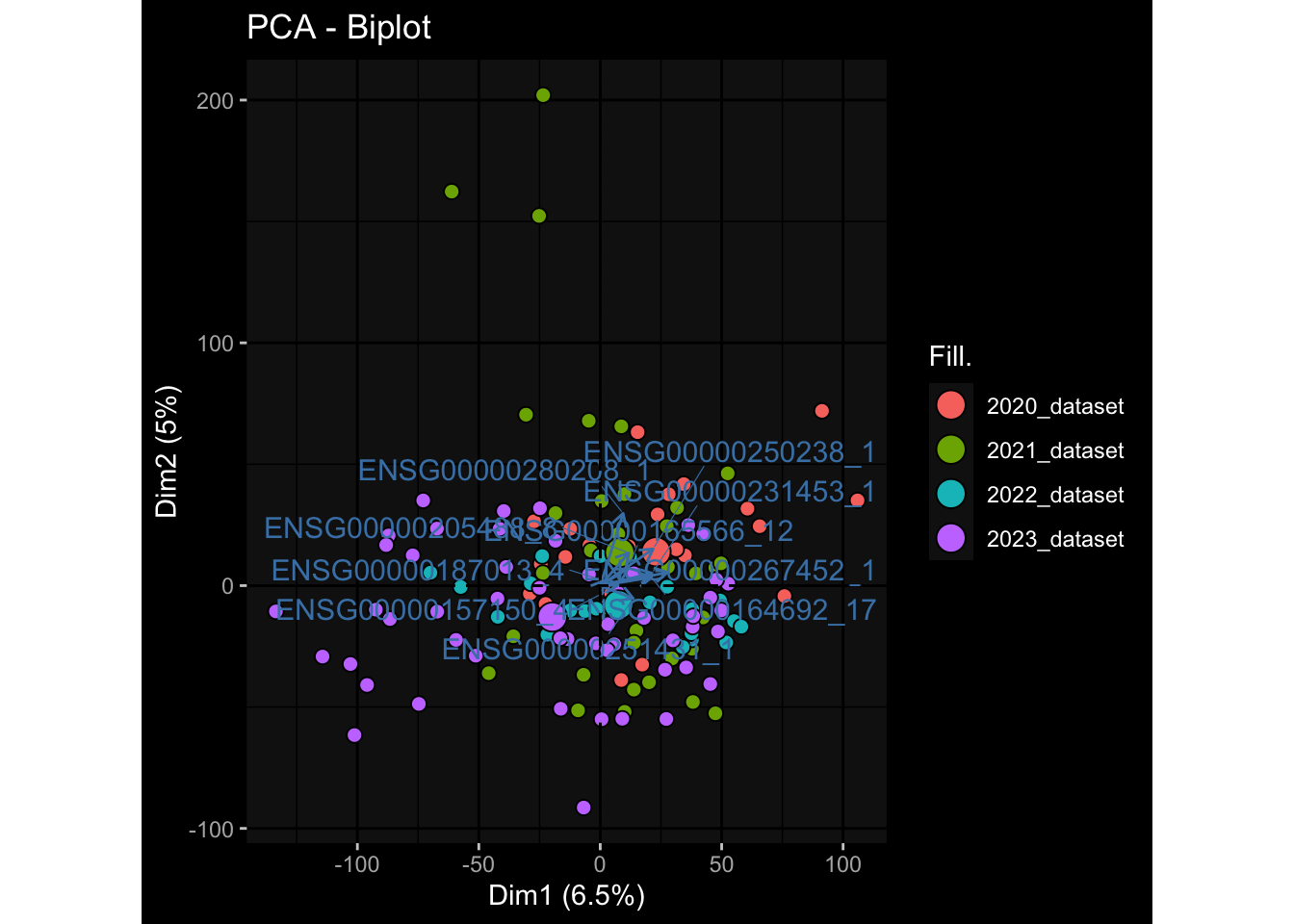
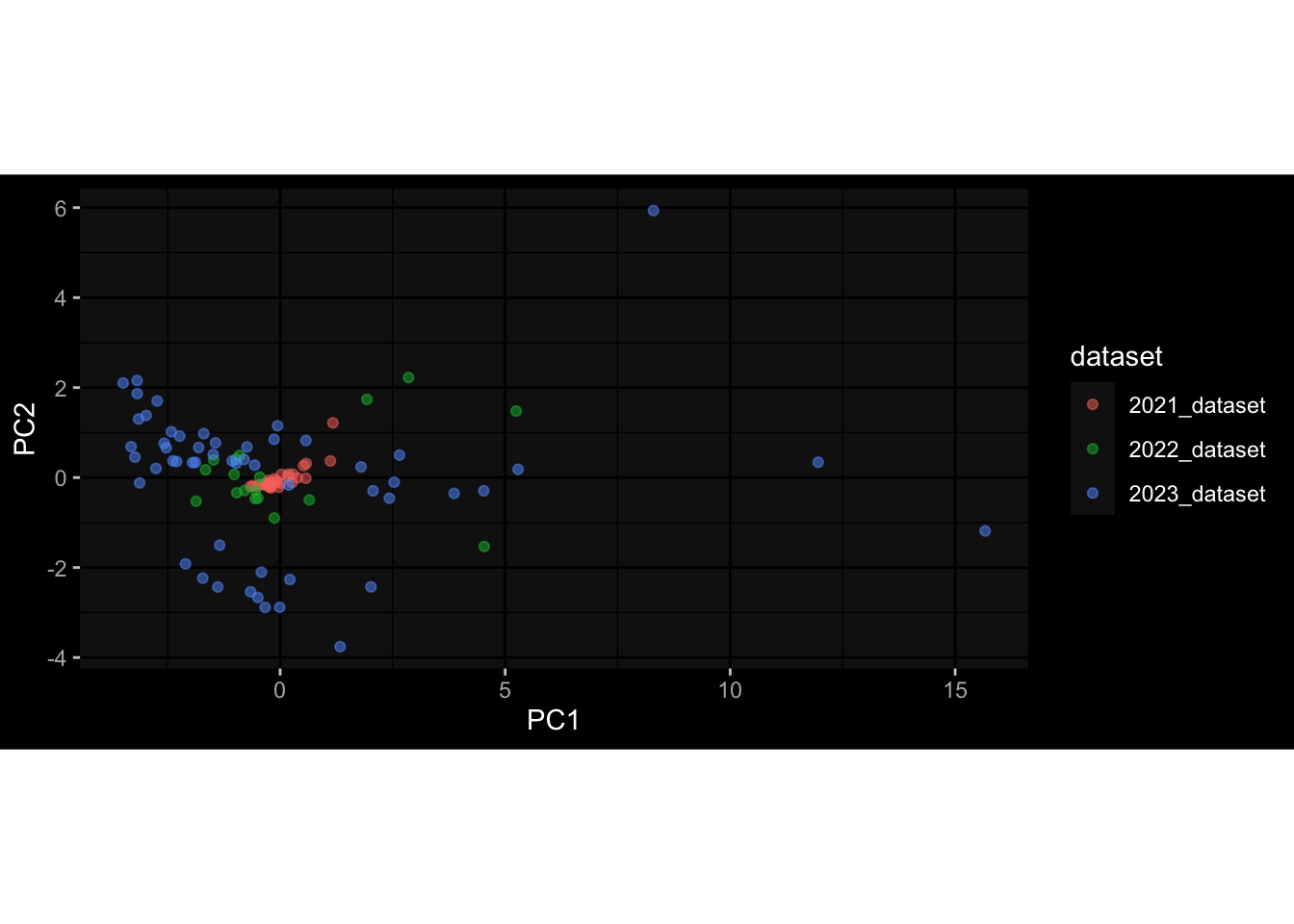
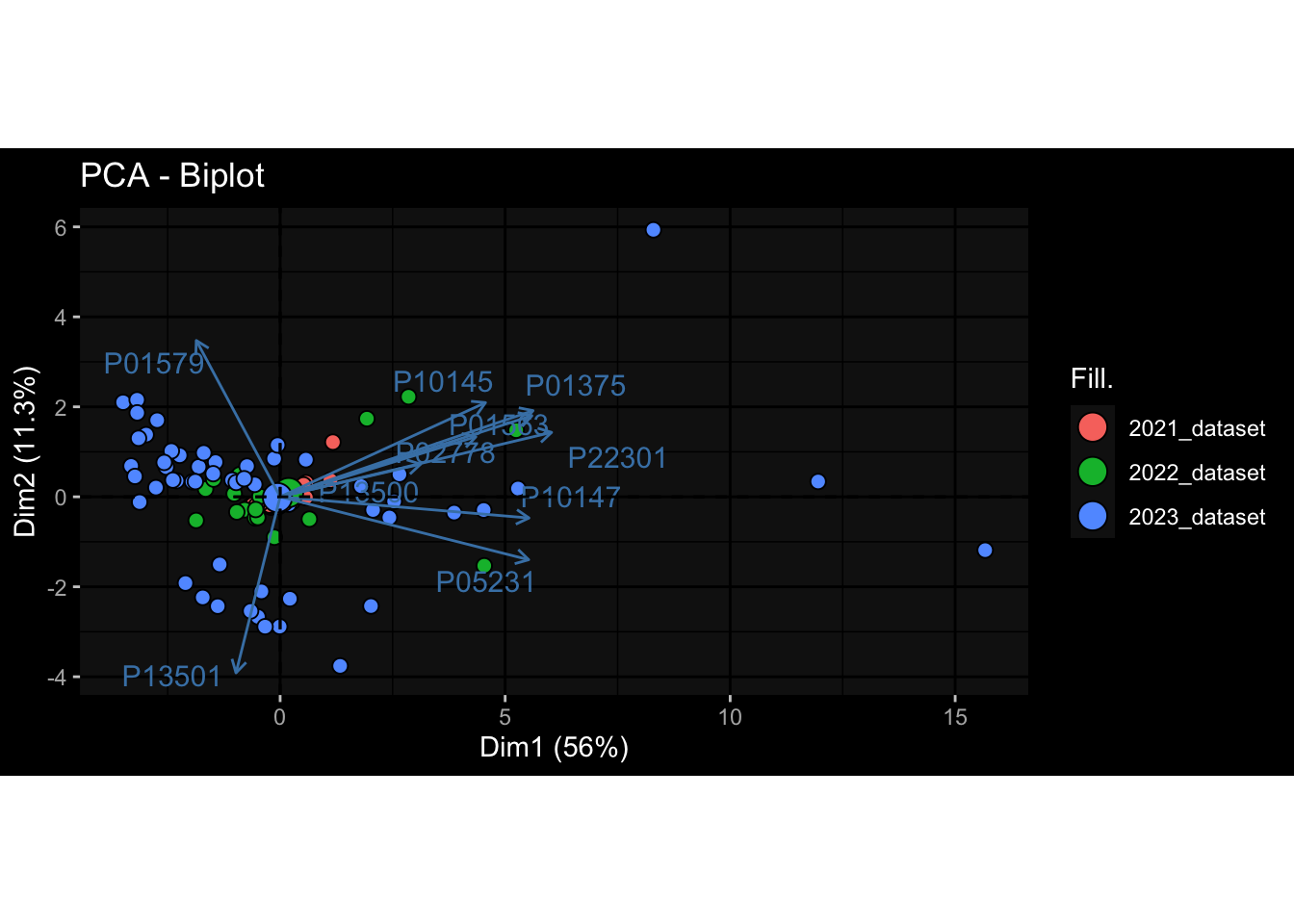
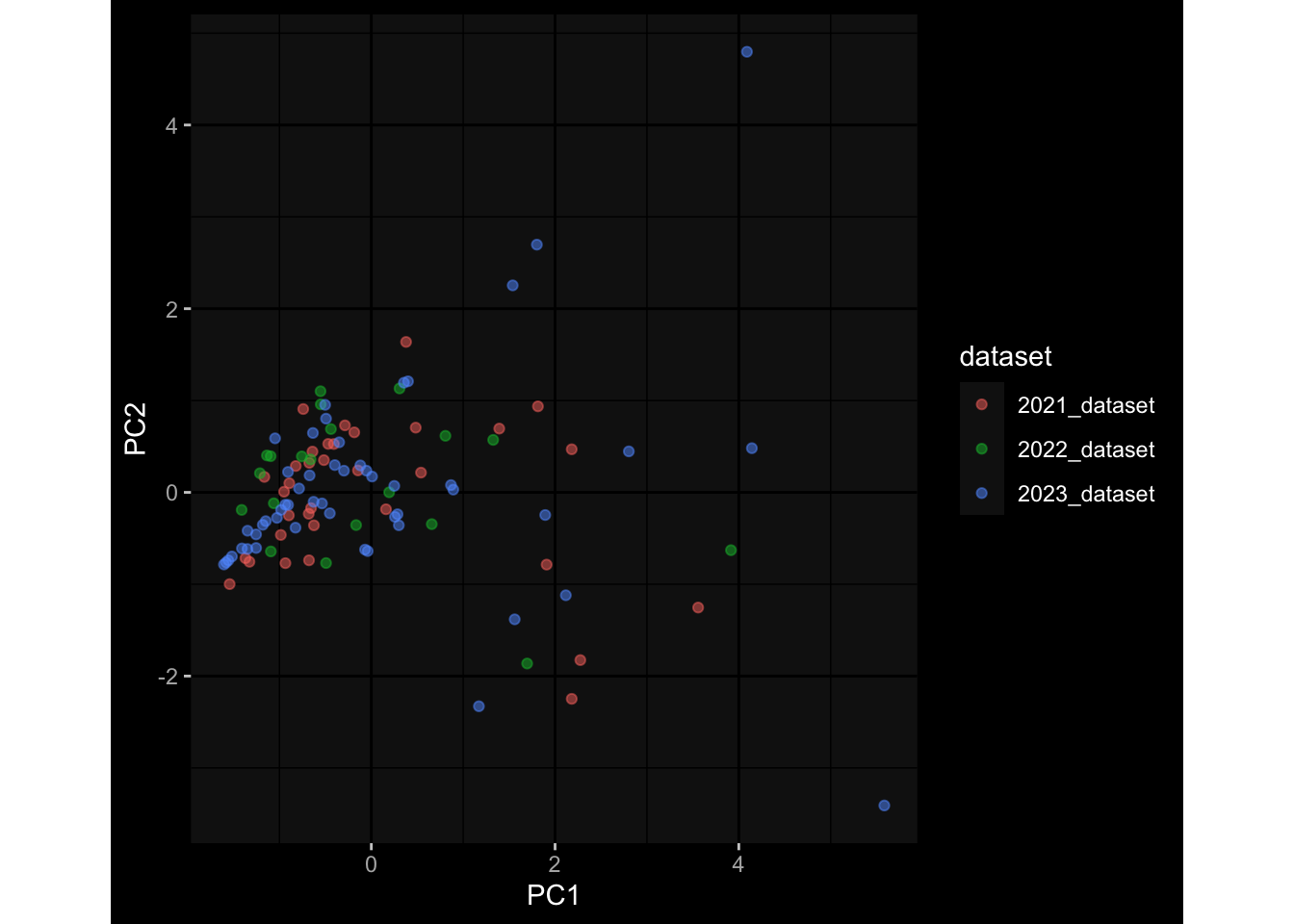
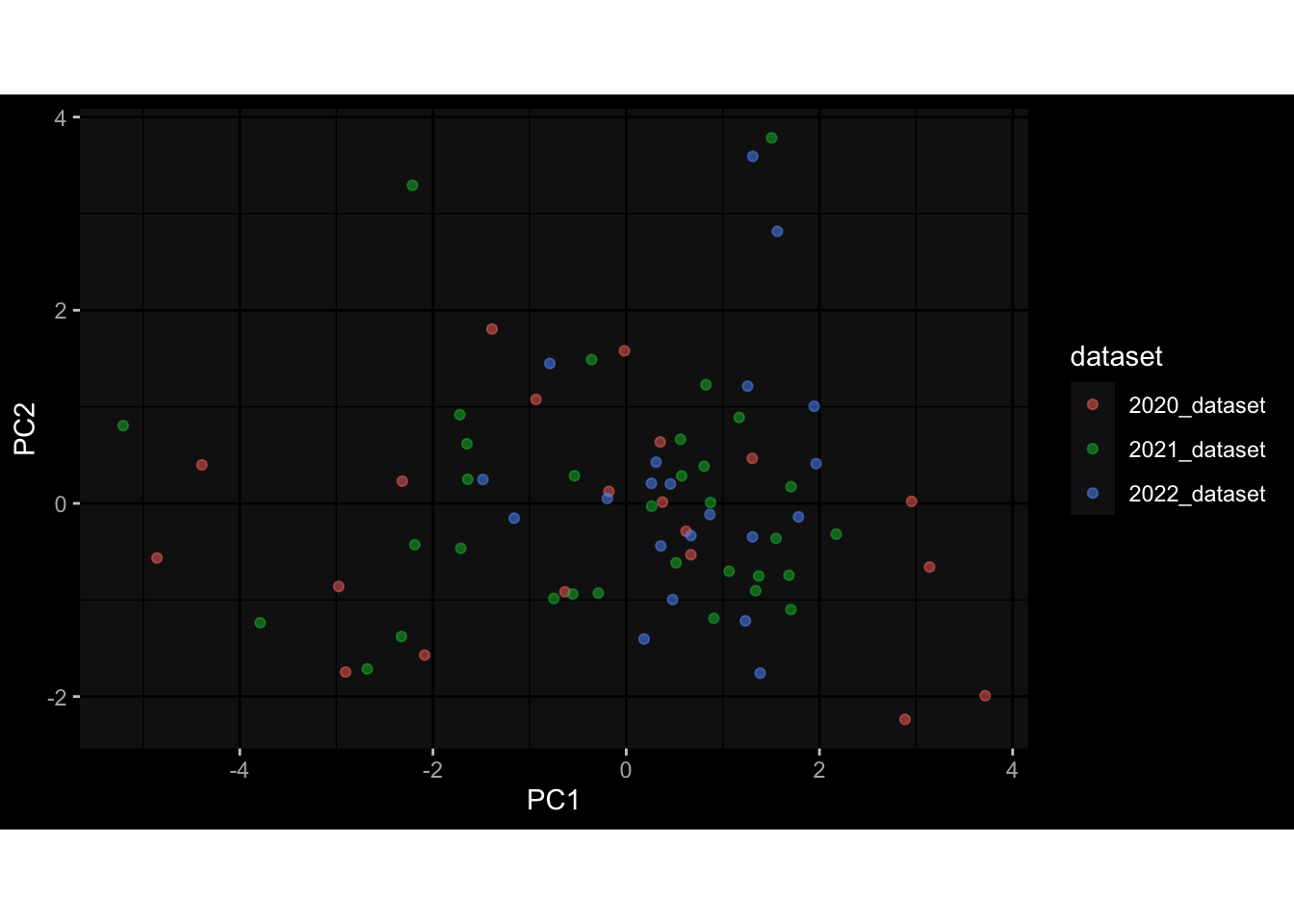
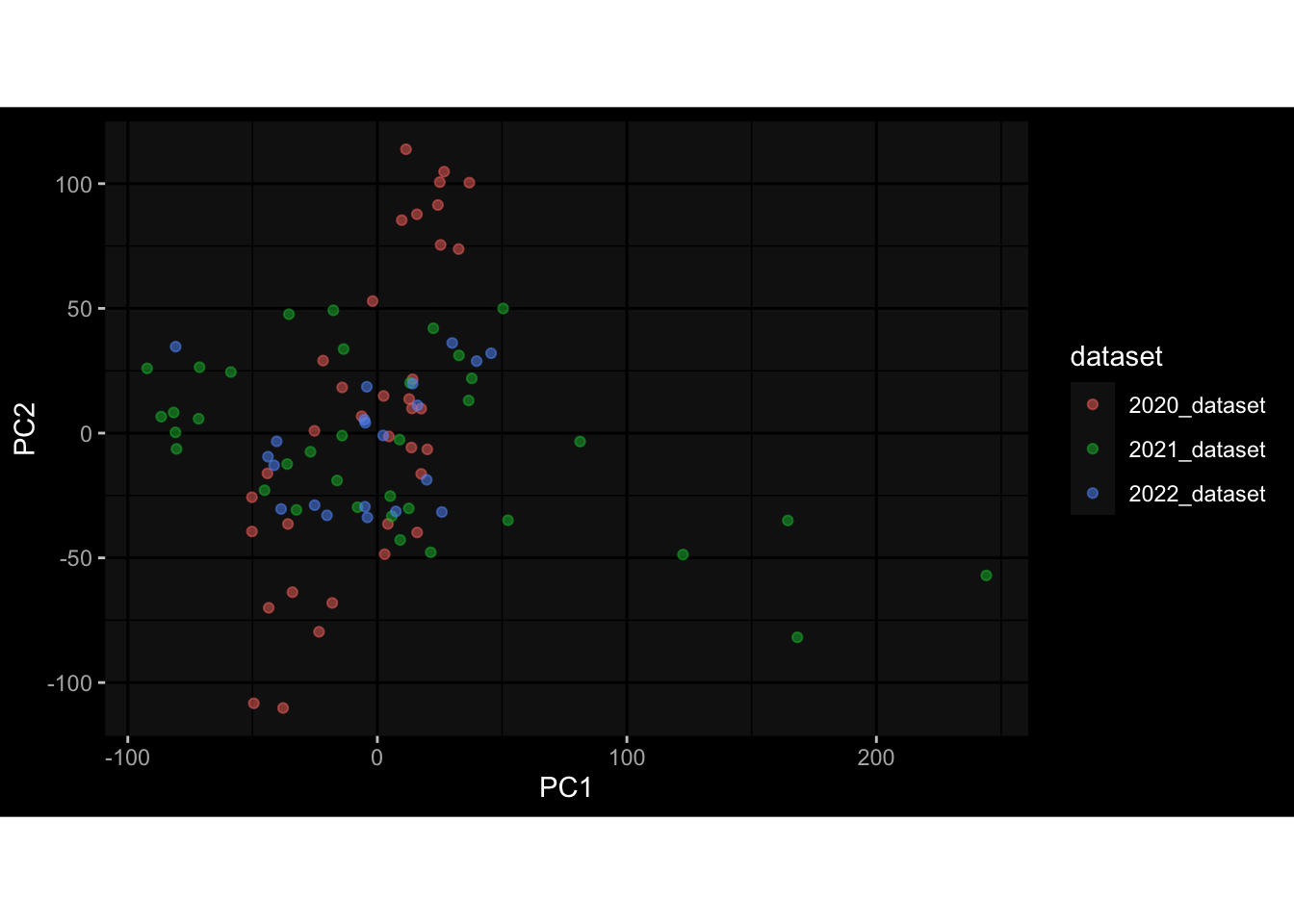
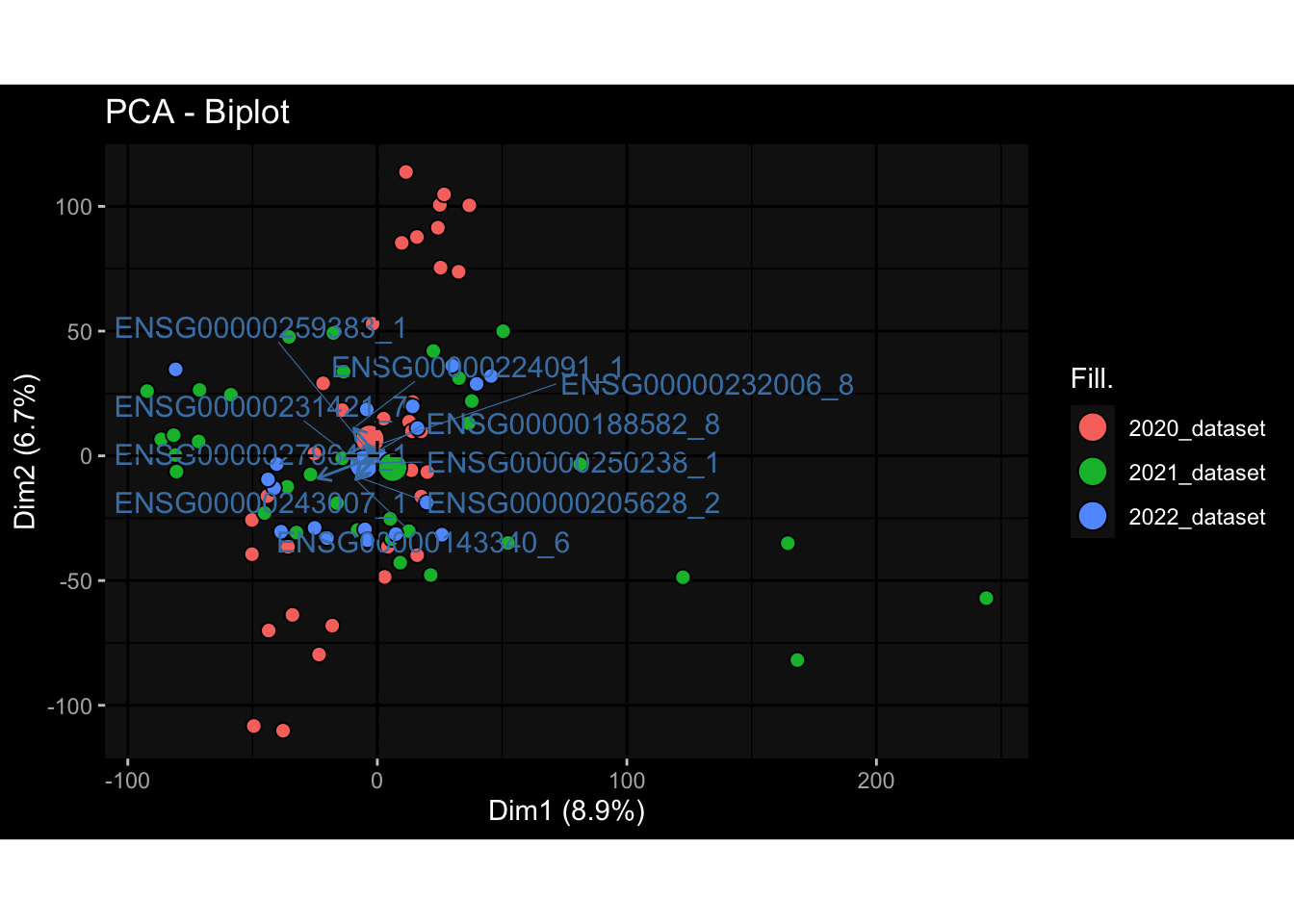
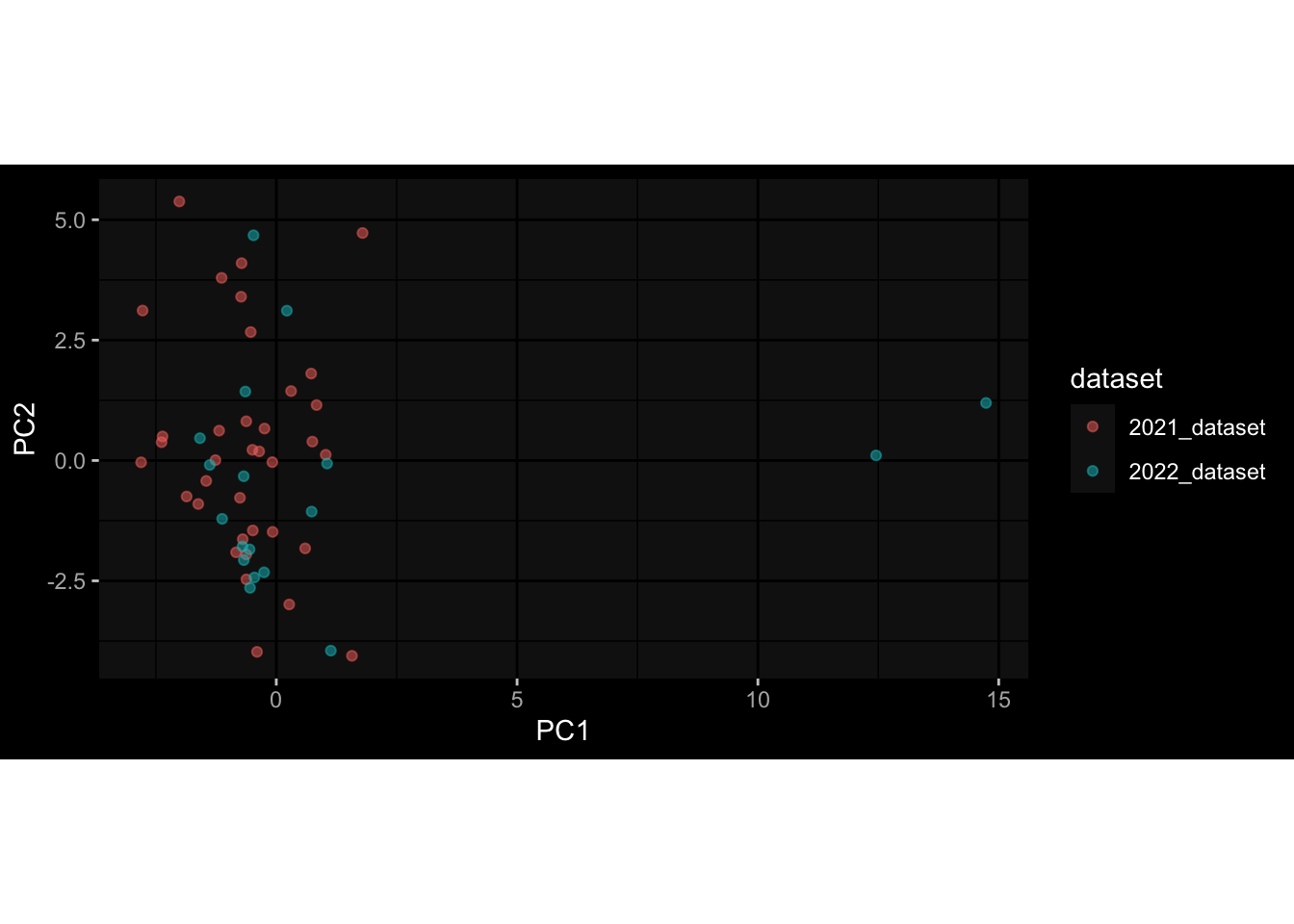
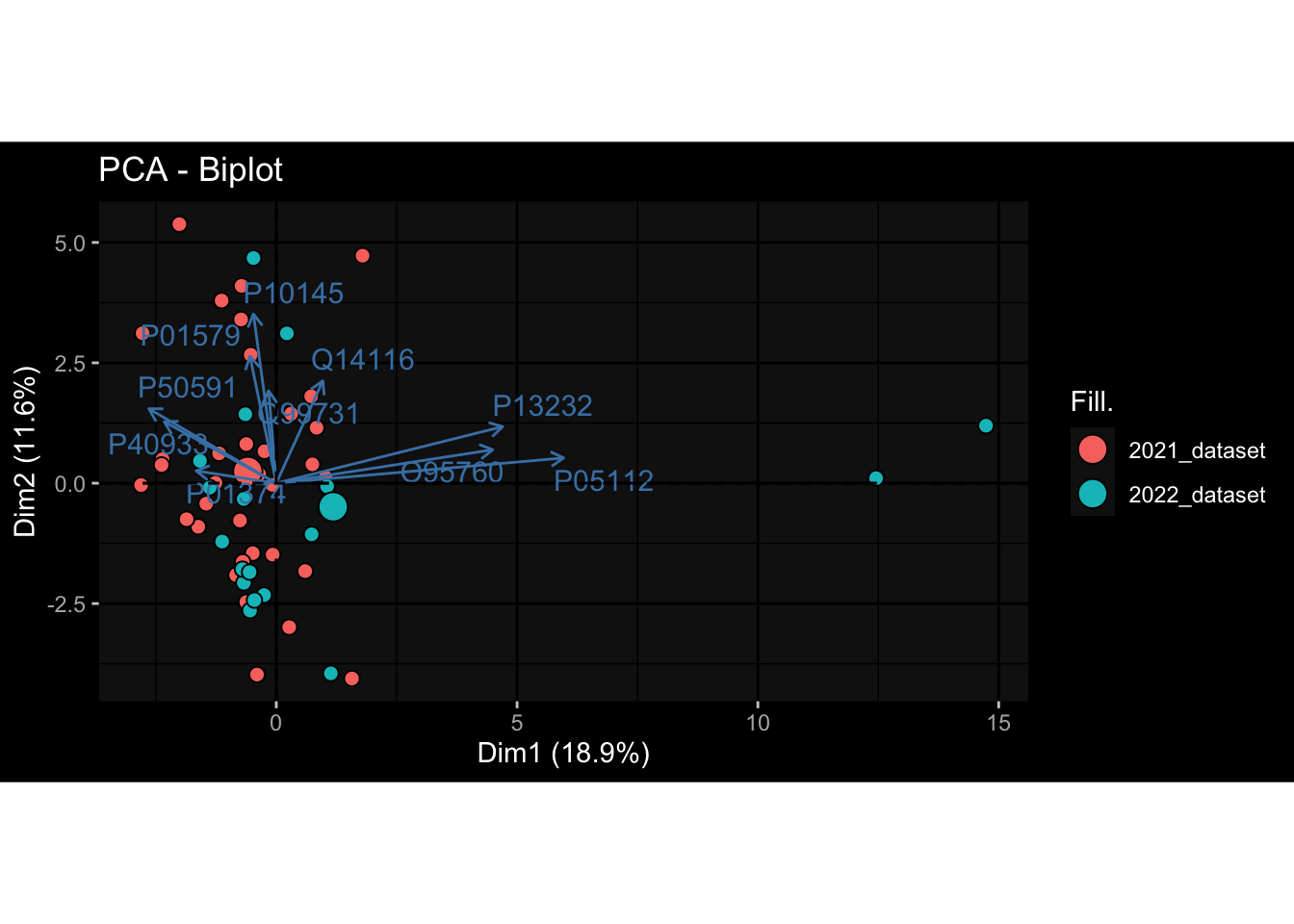
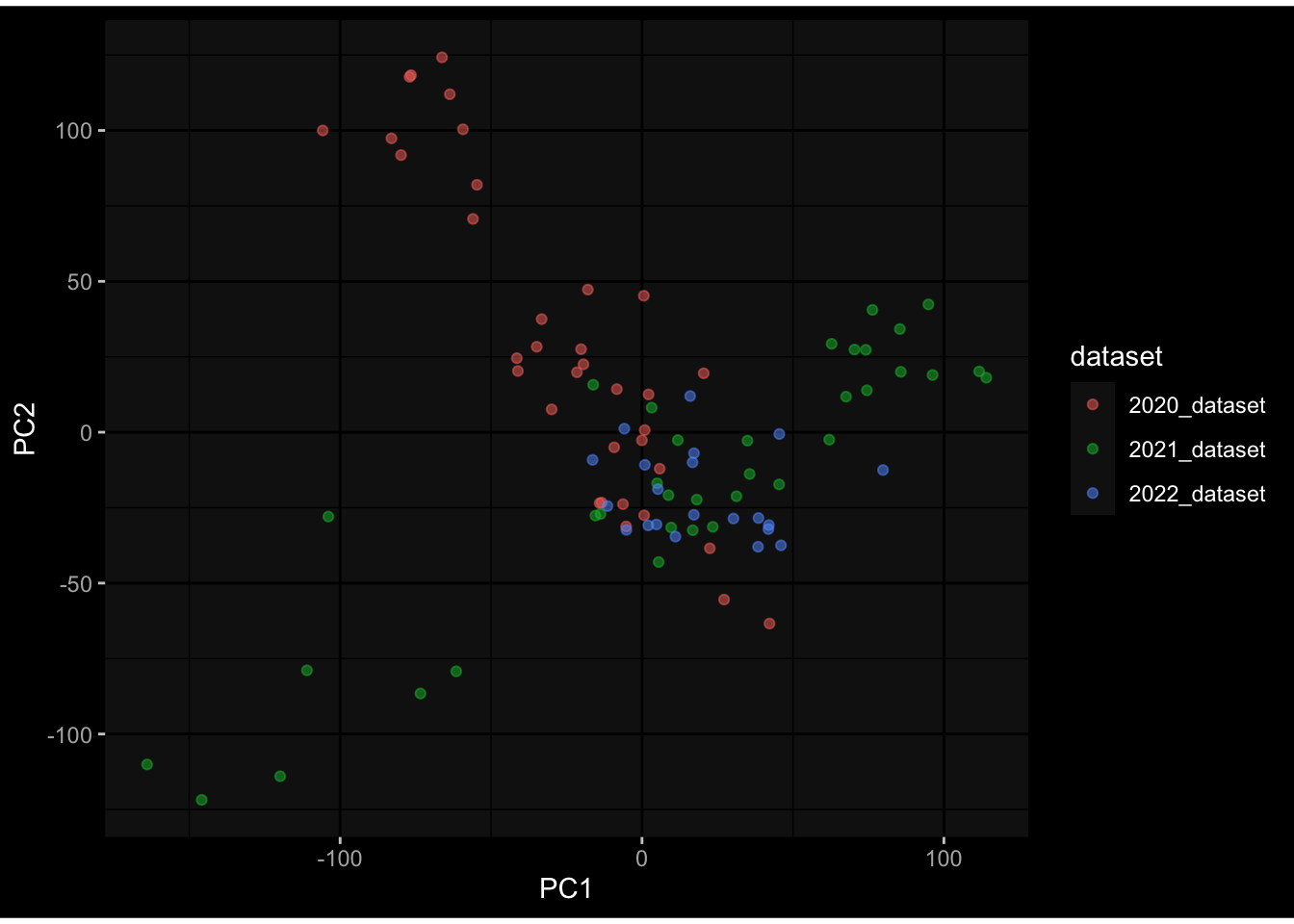
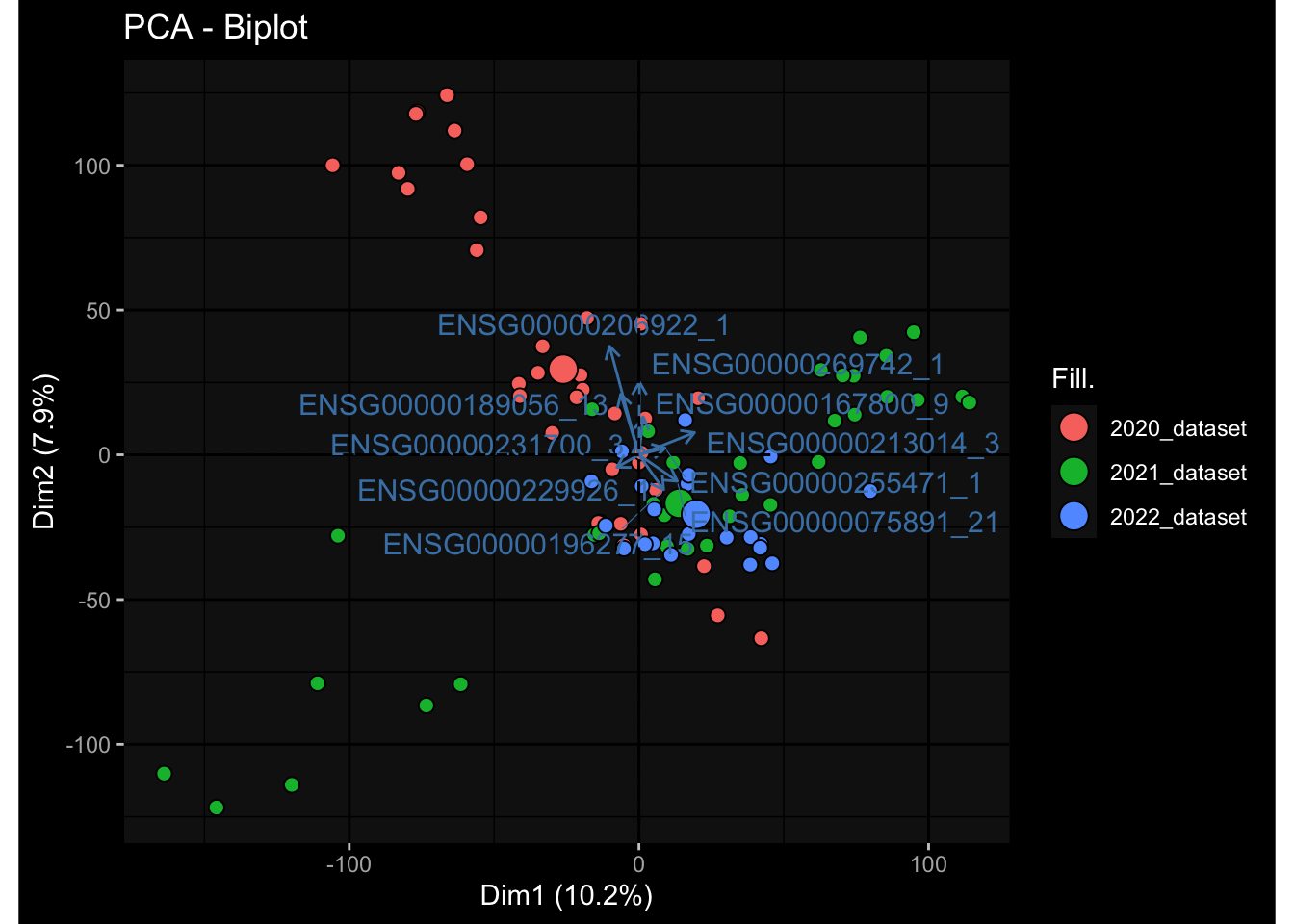
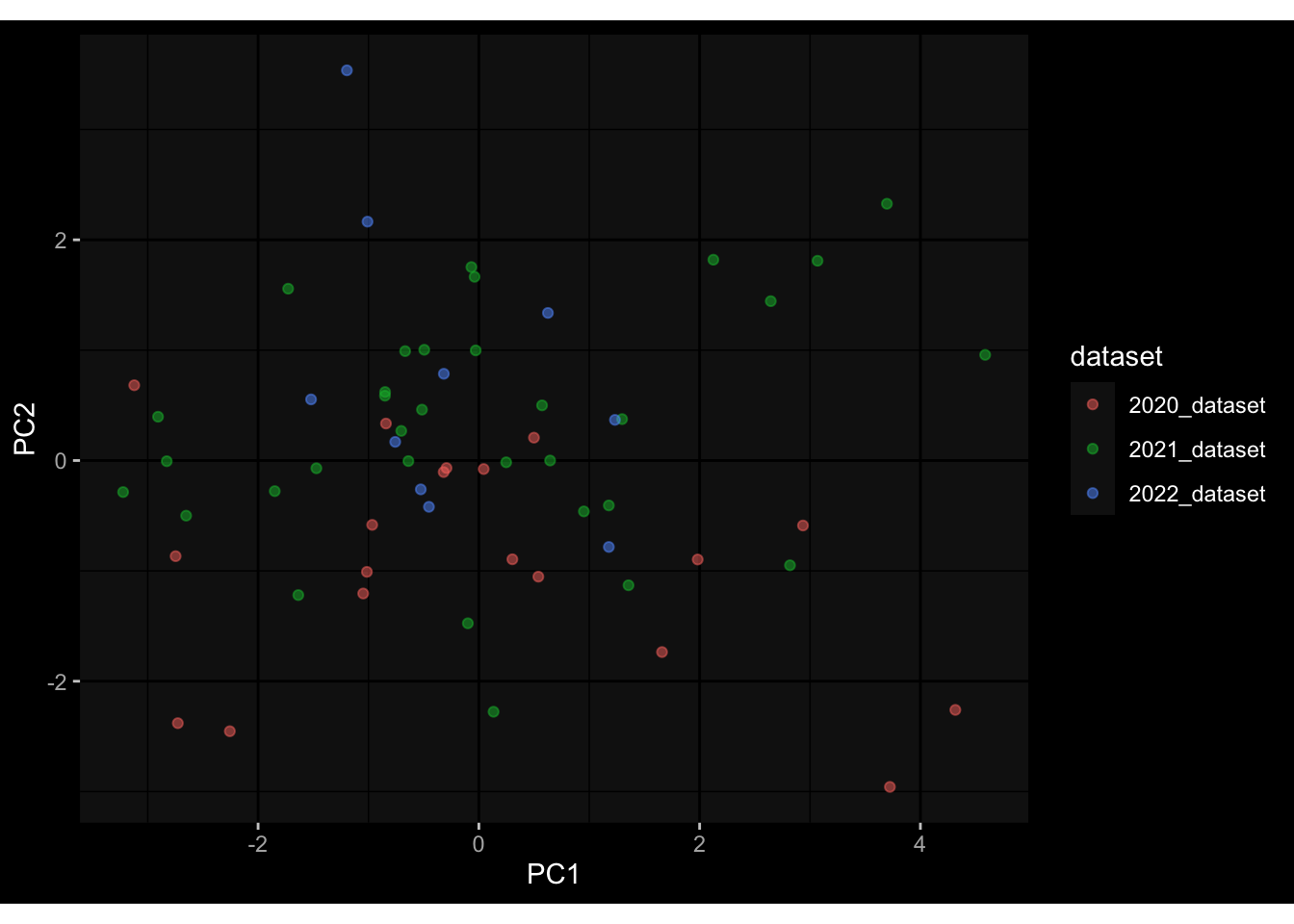
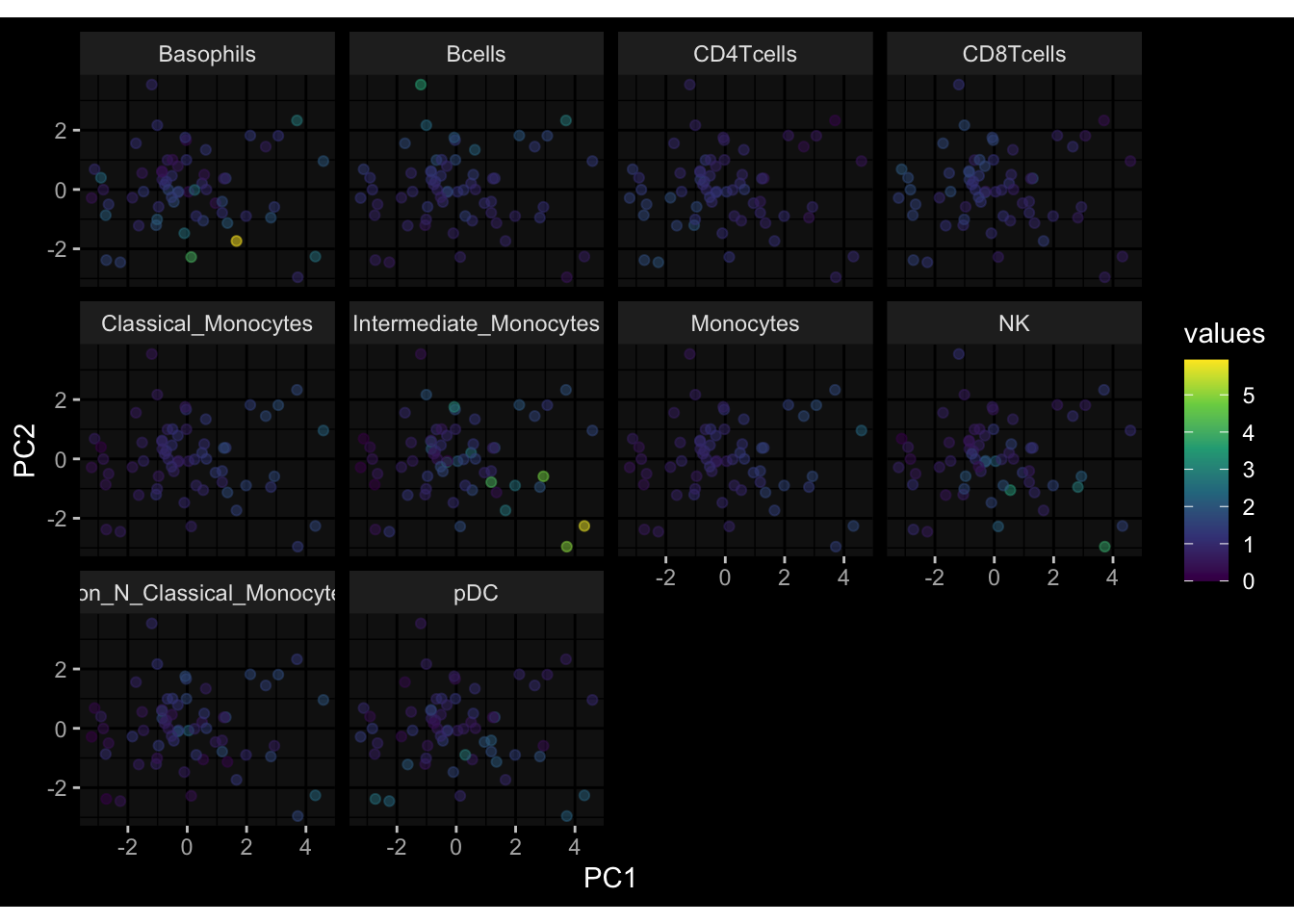
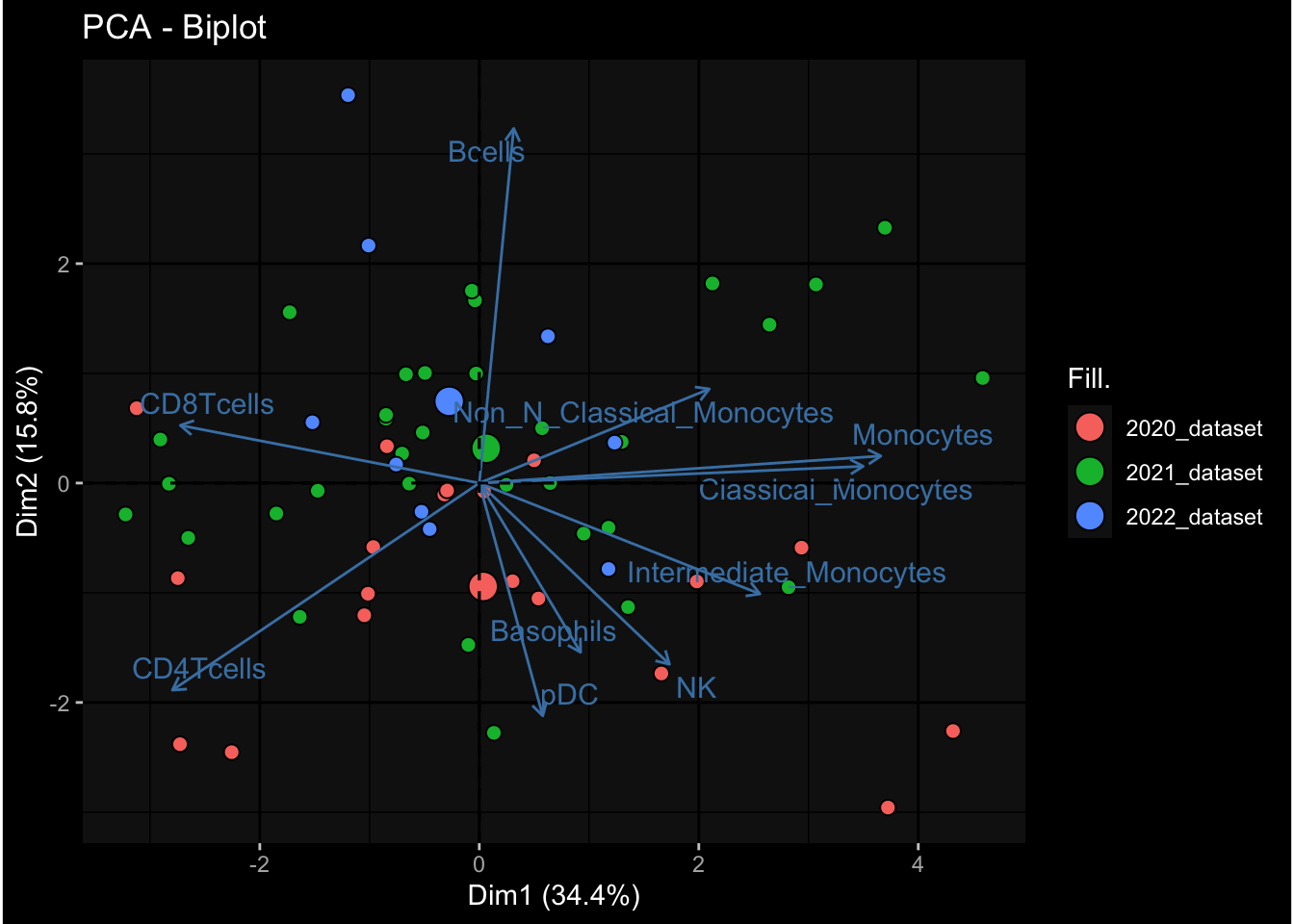
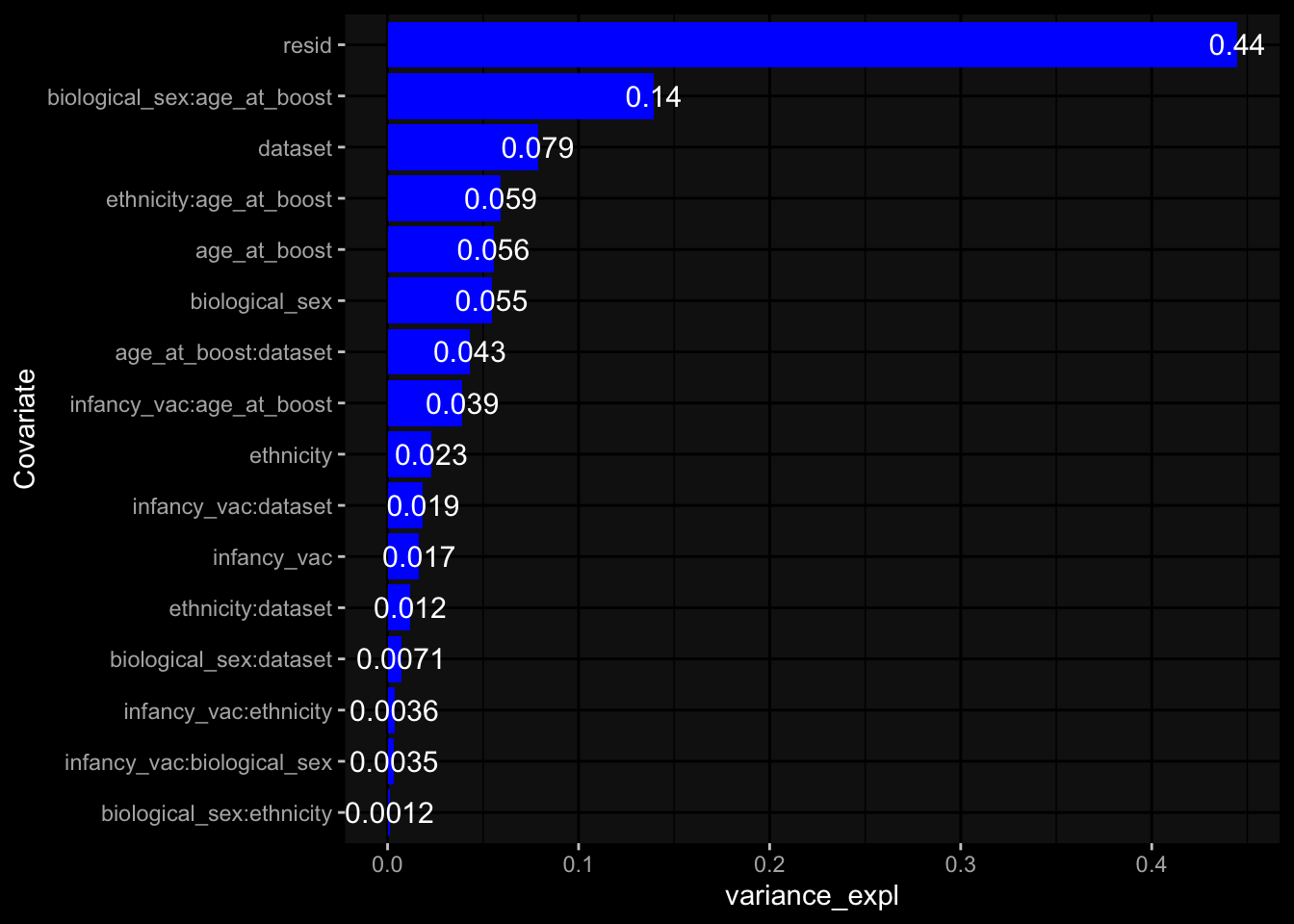
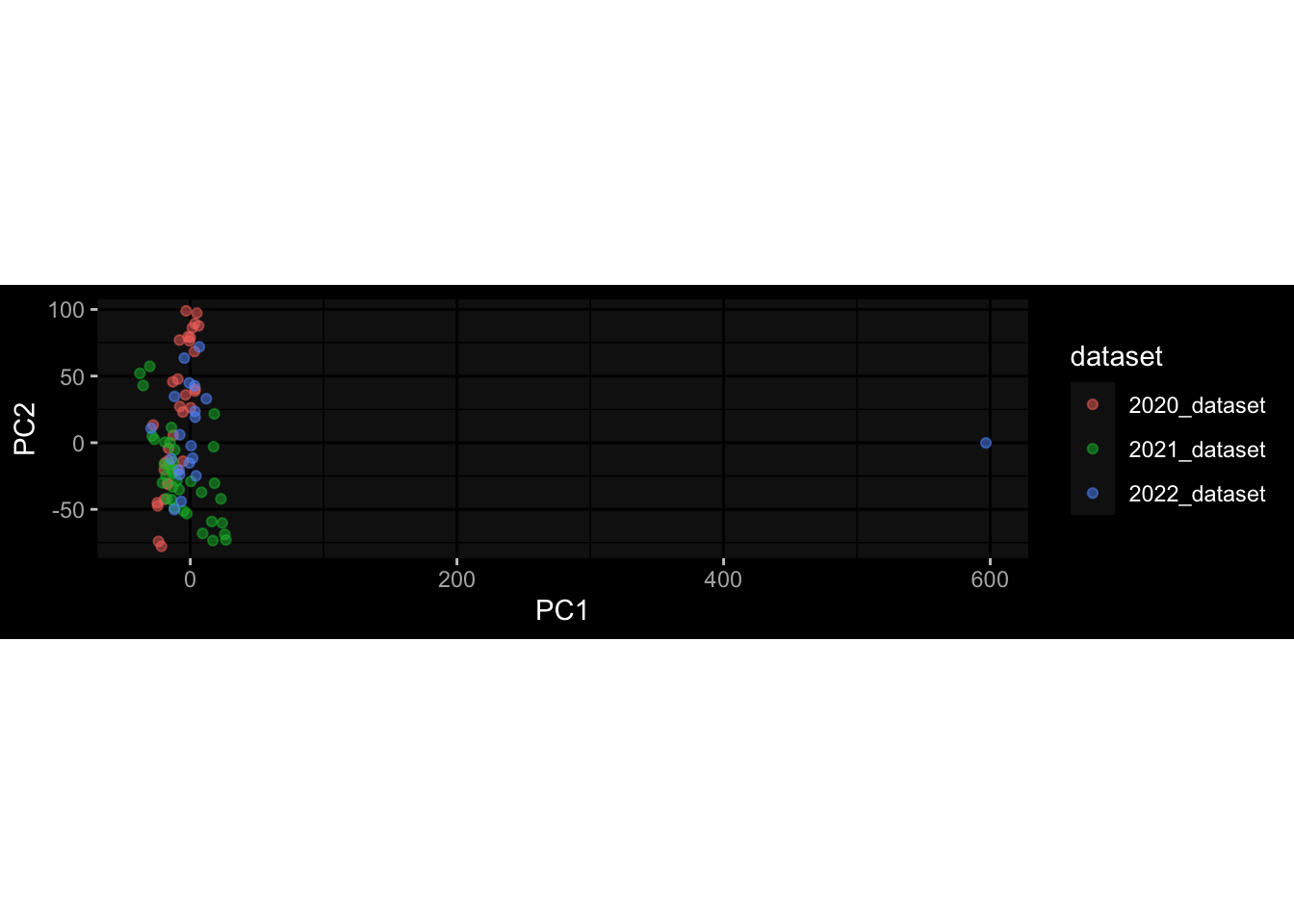
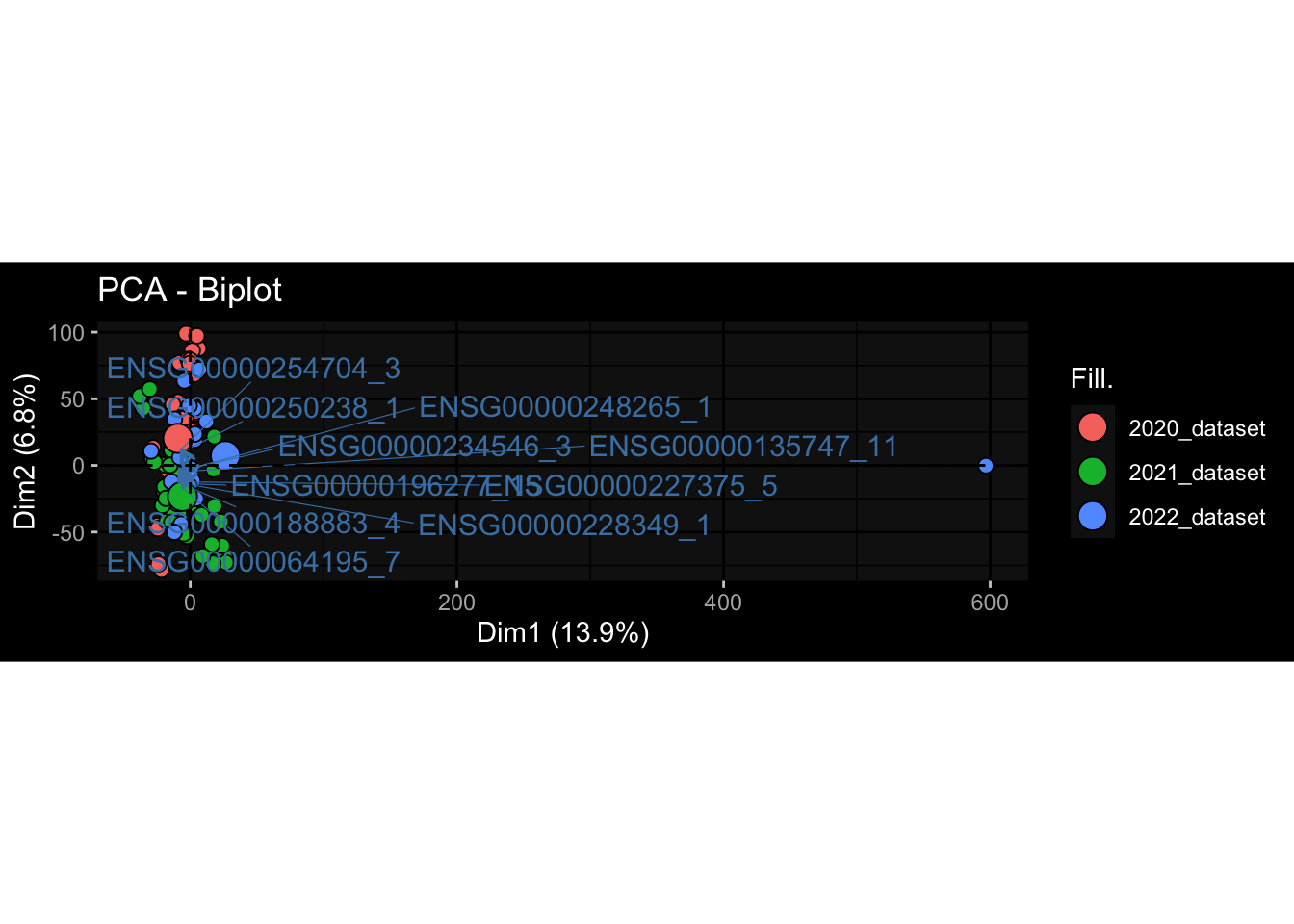
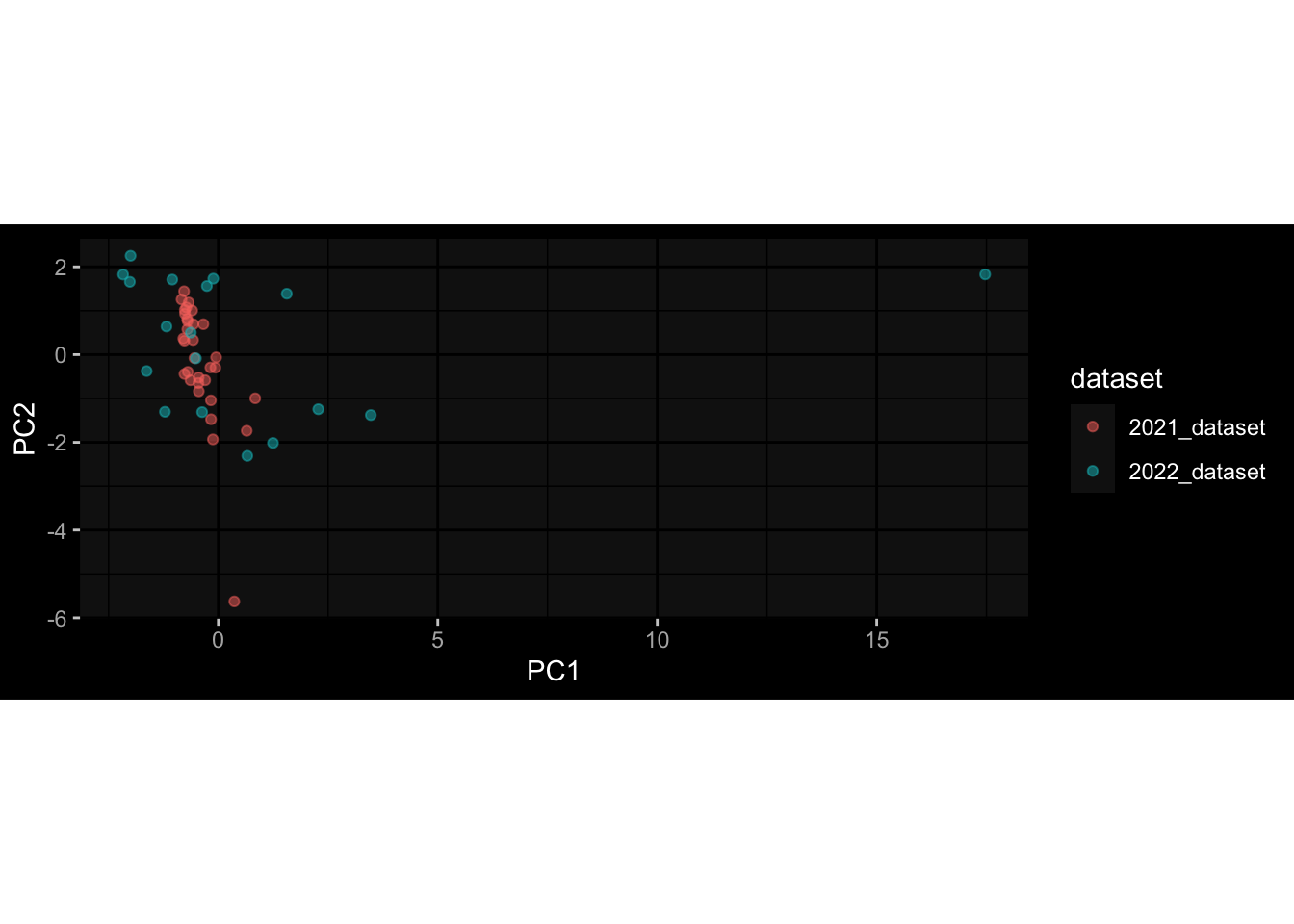
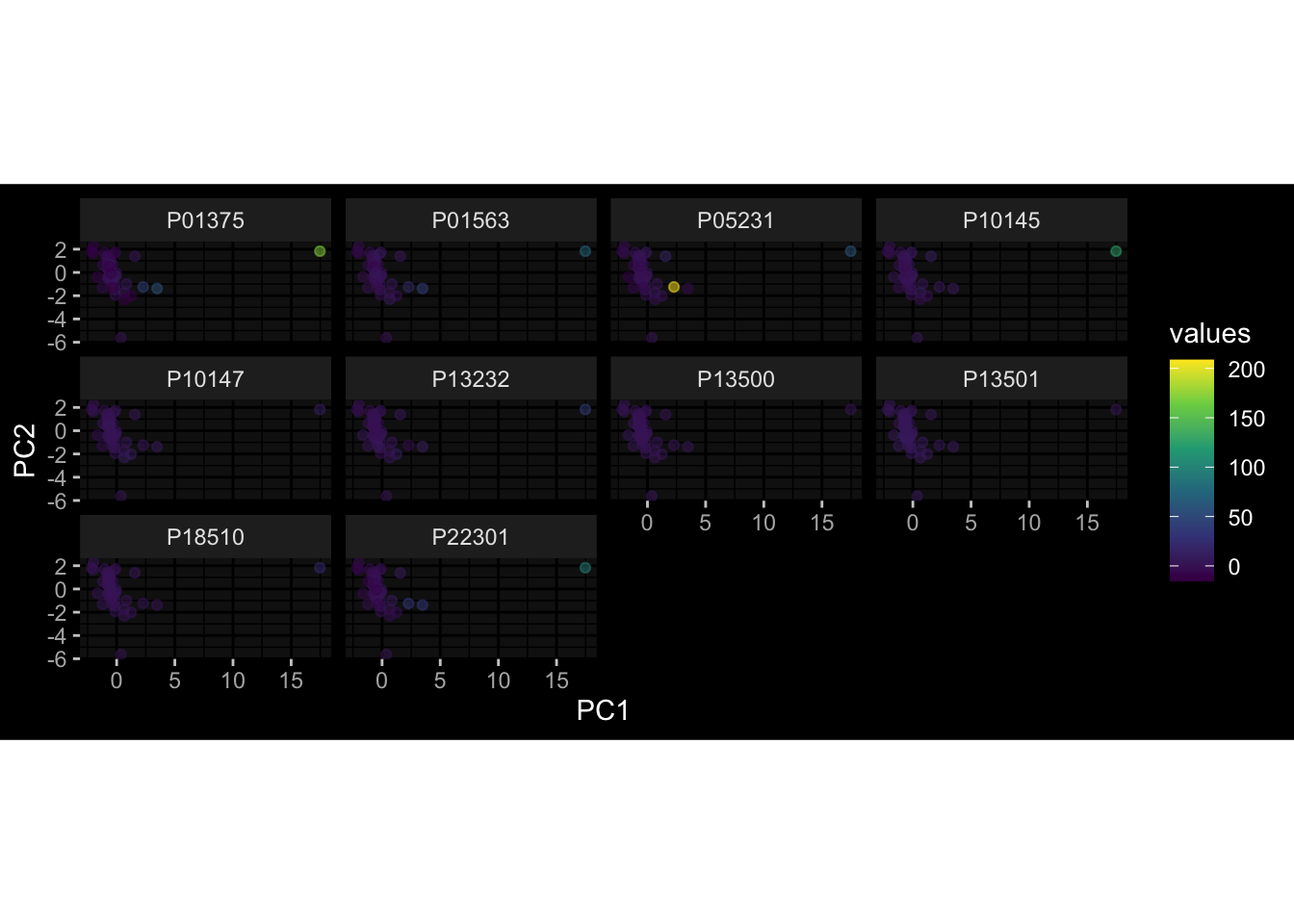
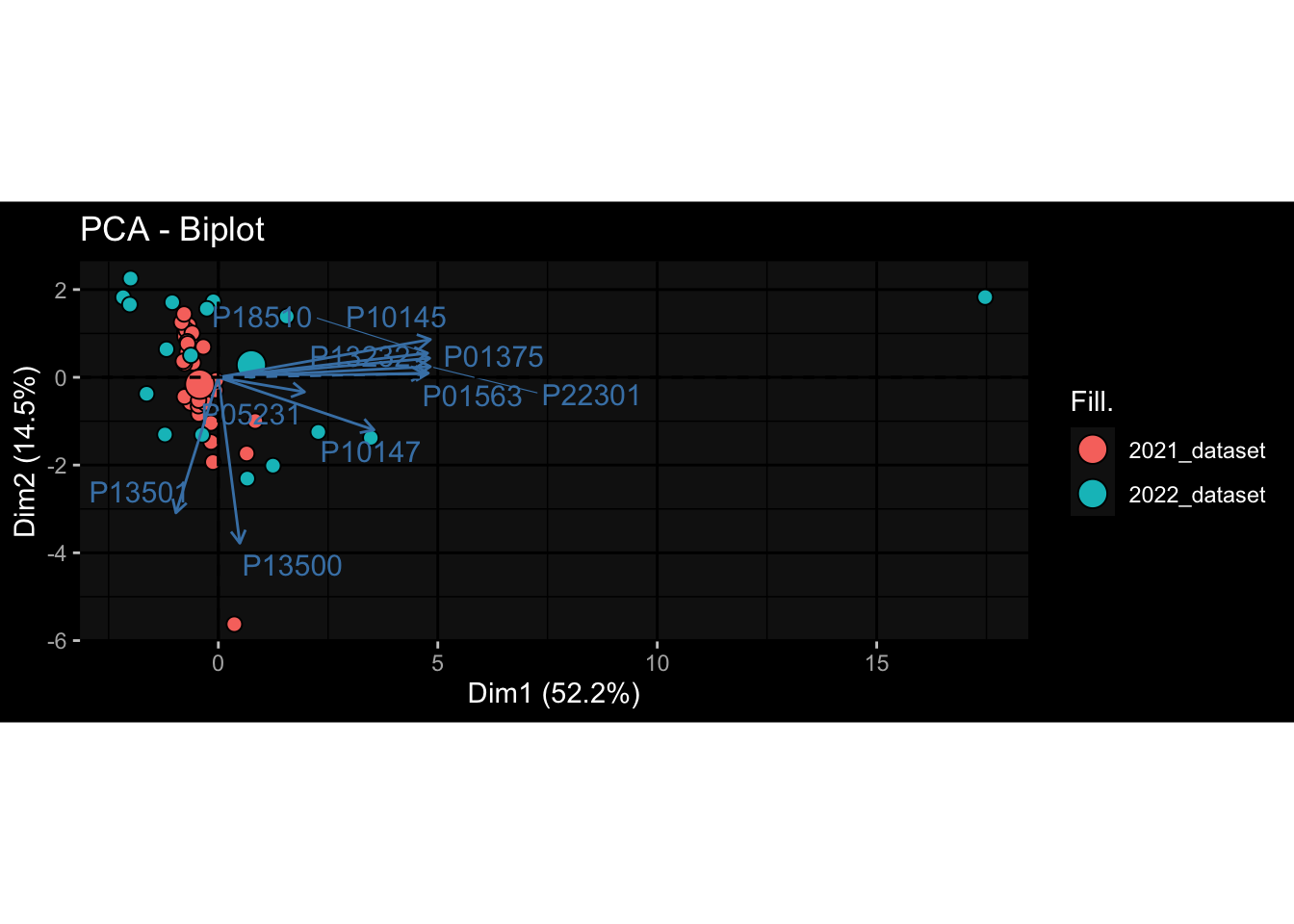
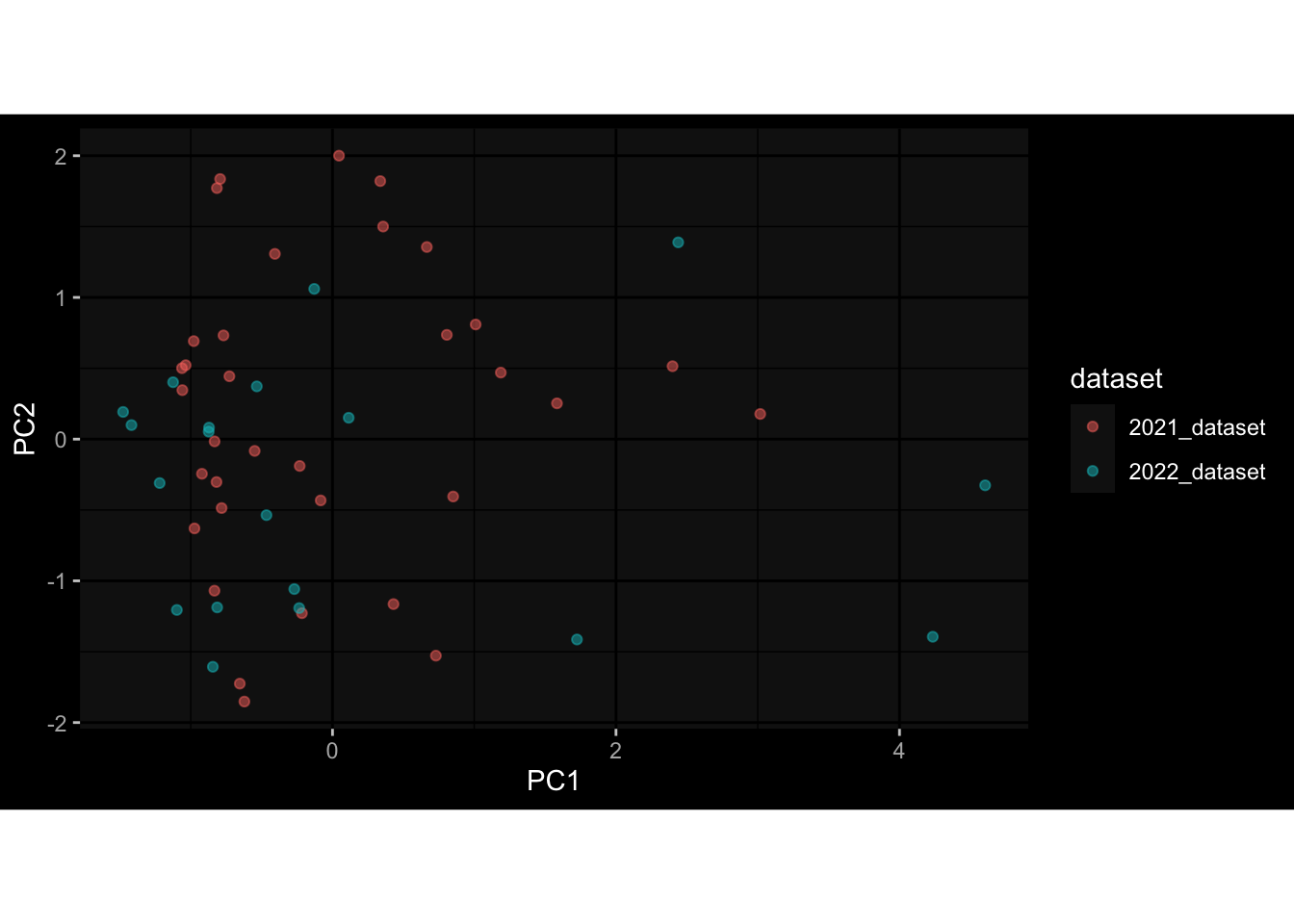
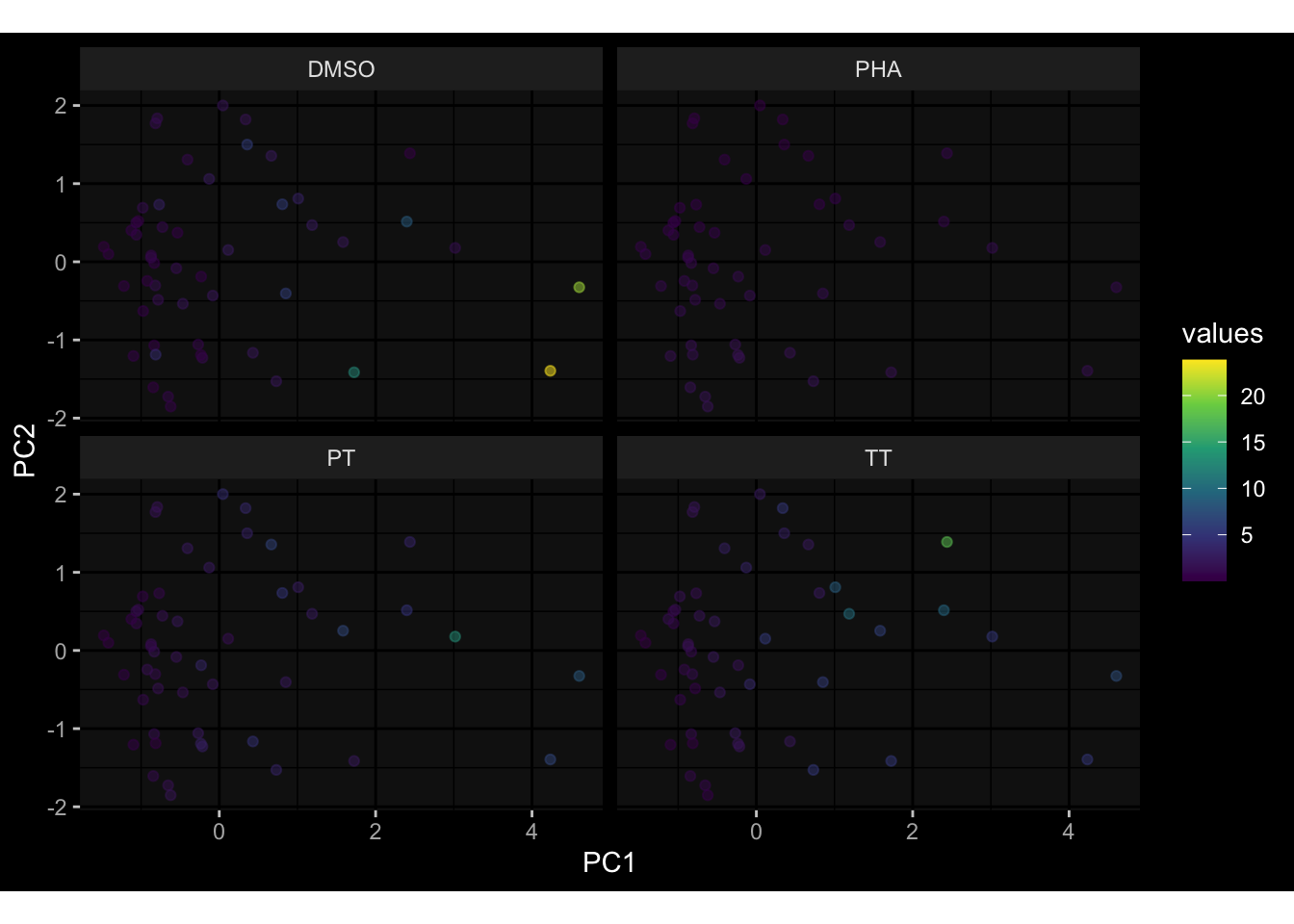
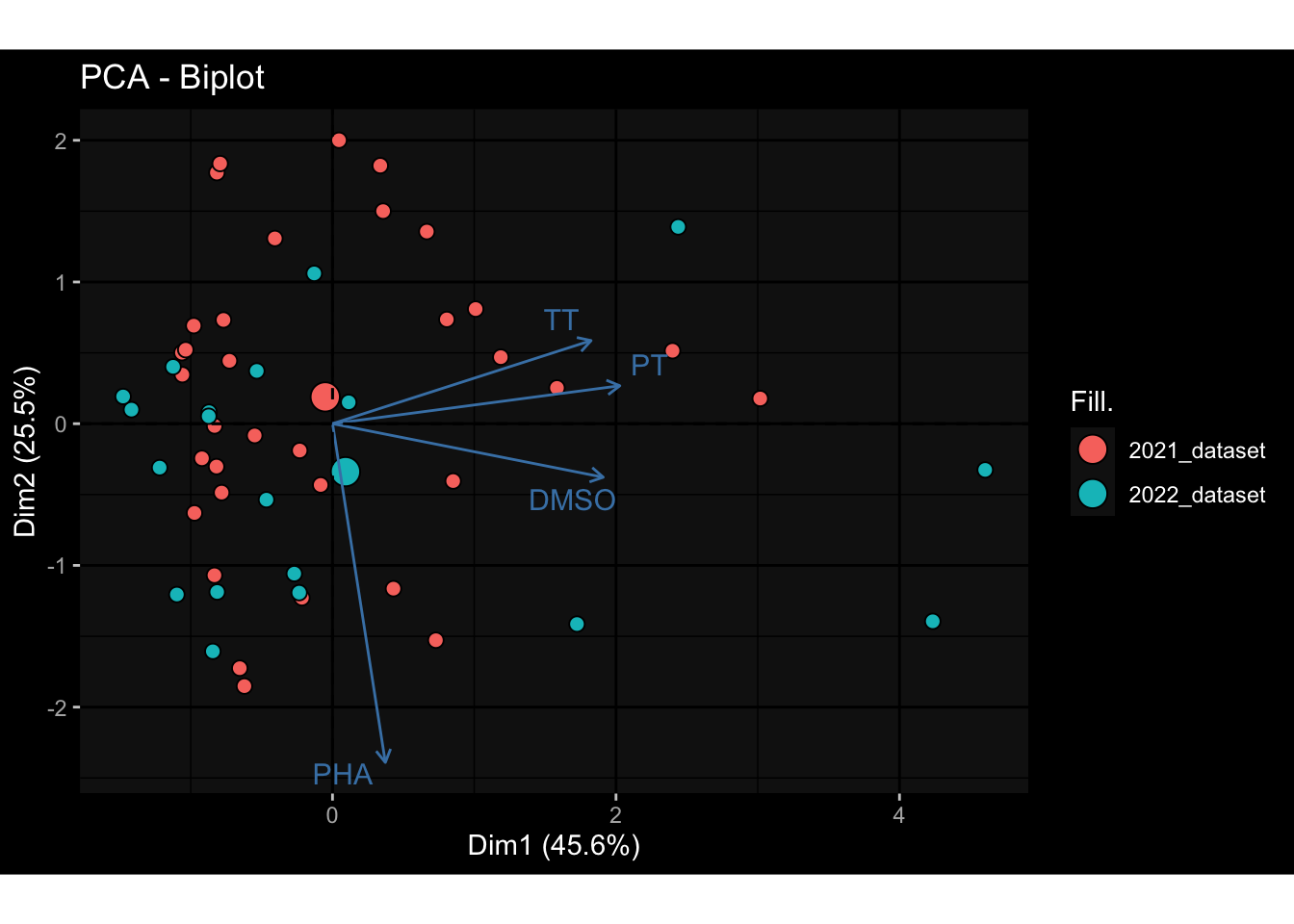
Even after running ComBat-seq there is still a substantial batch effect.
However, here https://rpubs.com/pshinde/Alldata_batcheffect_correction_3rd_challenge, there is not batch effect, I am not sure why that is.
Maybe it is because I am batch-correcting the data for each day relative to boost?
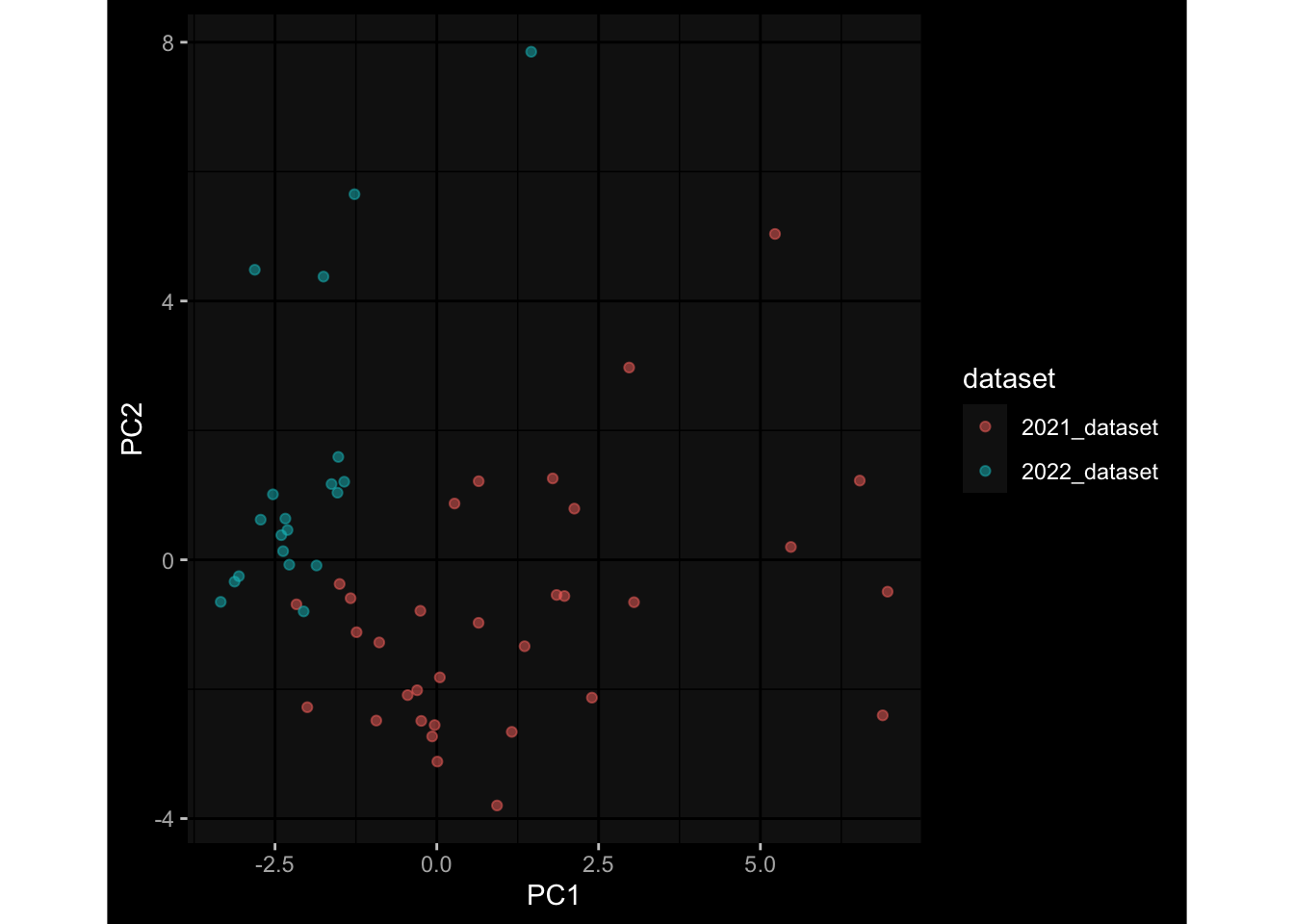
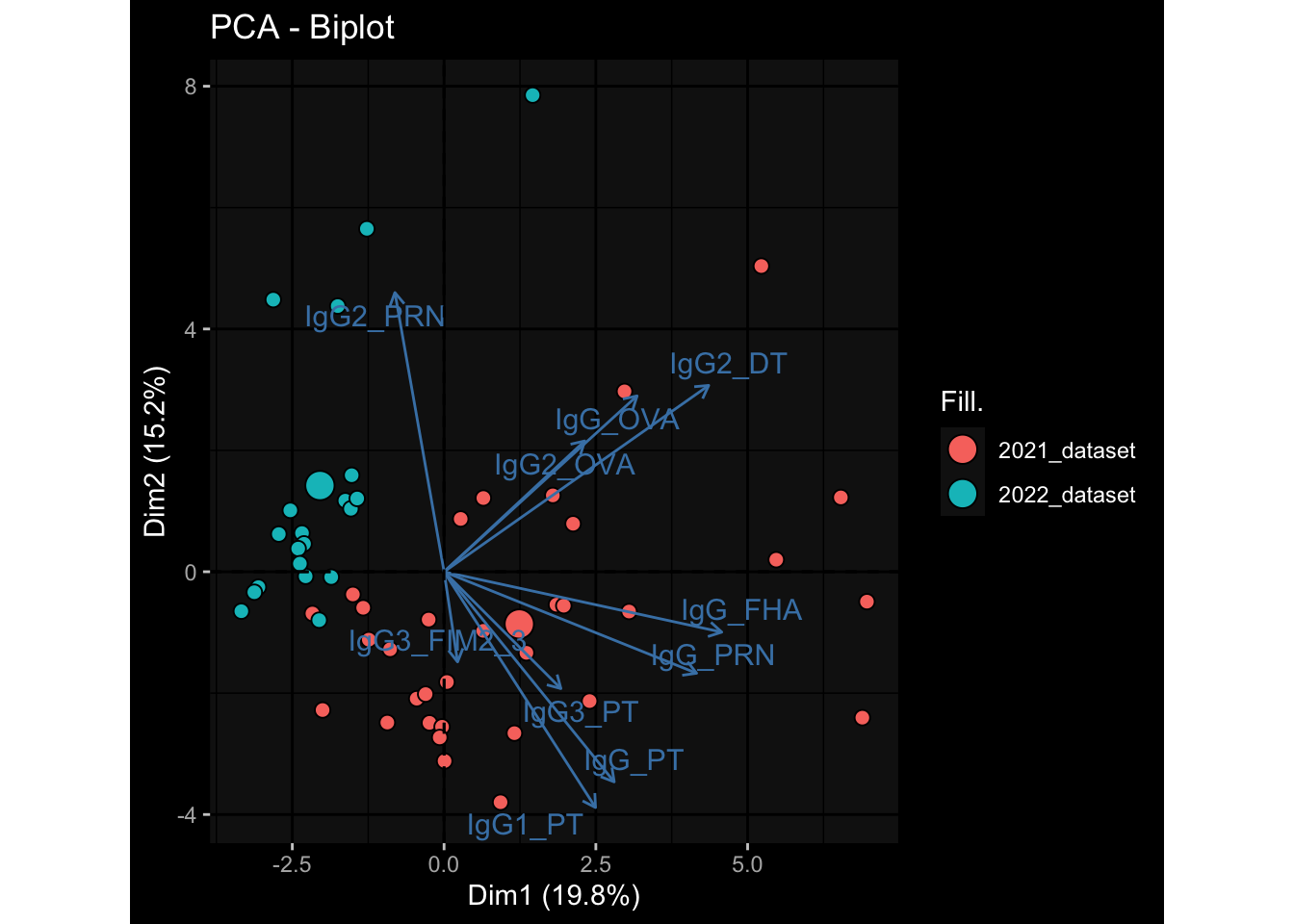
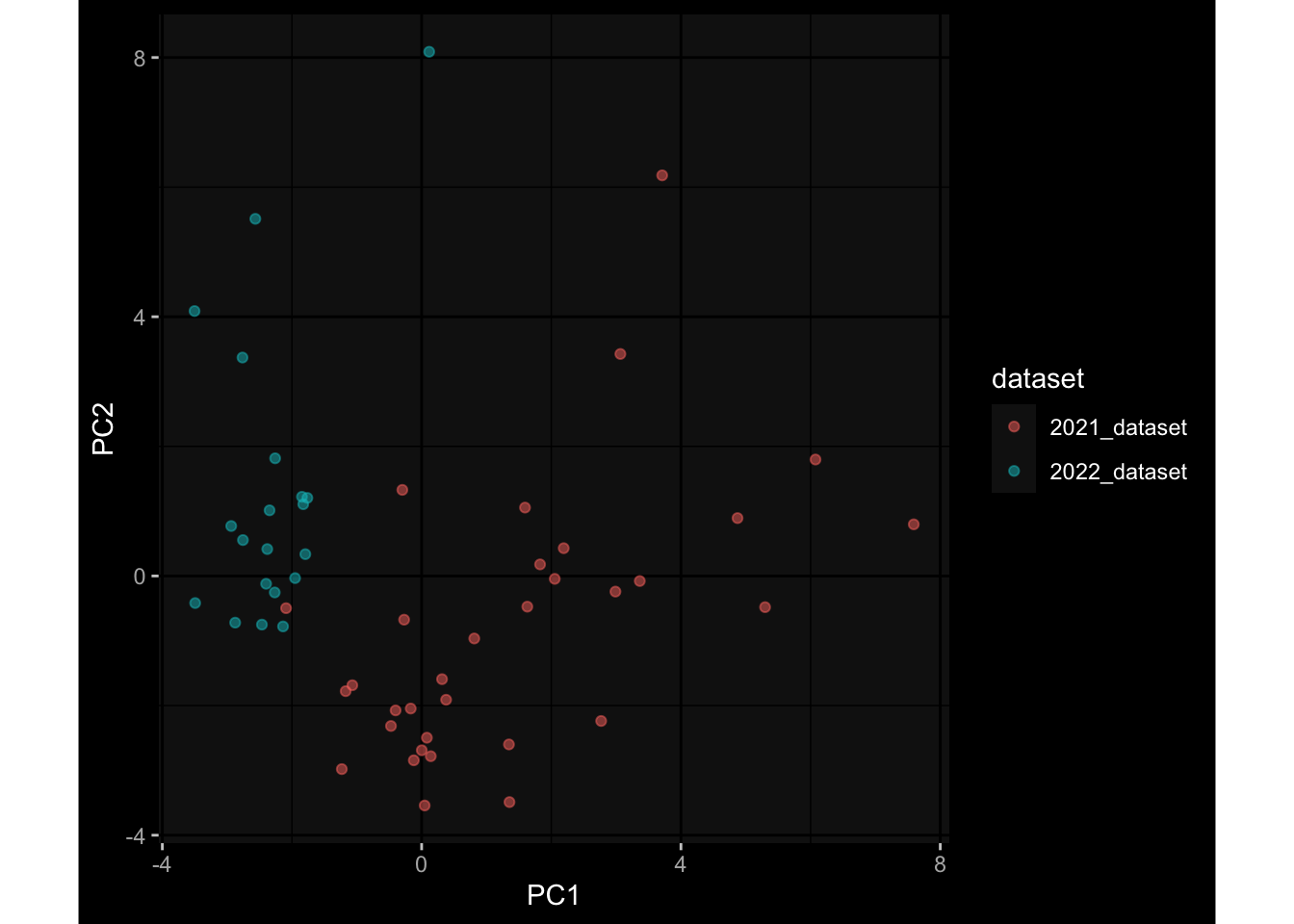
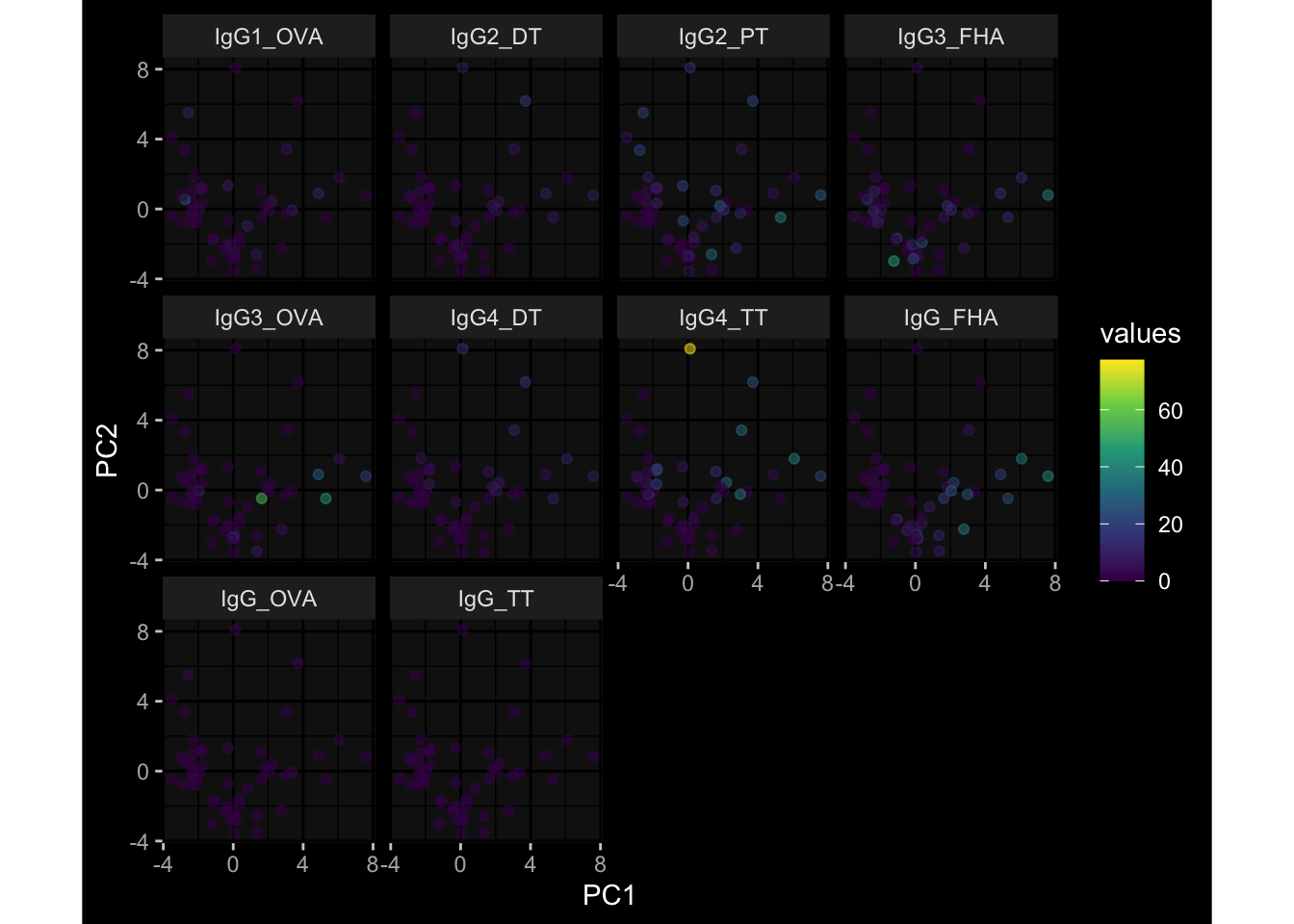
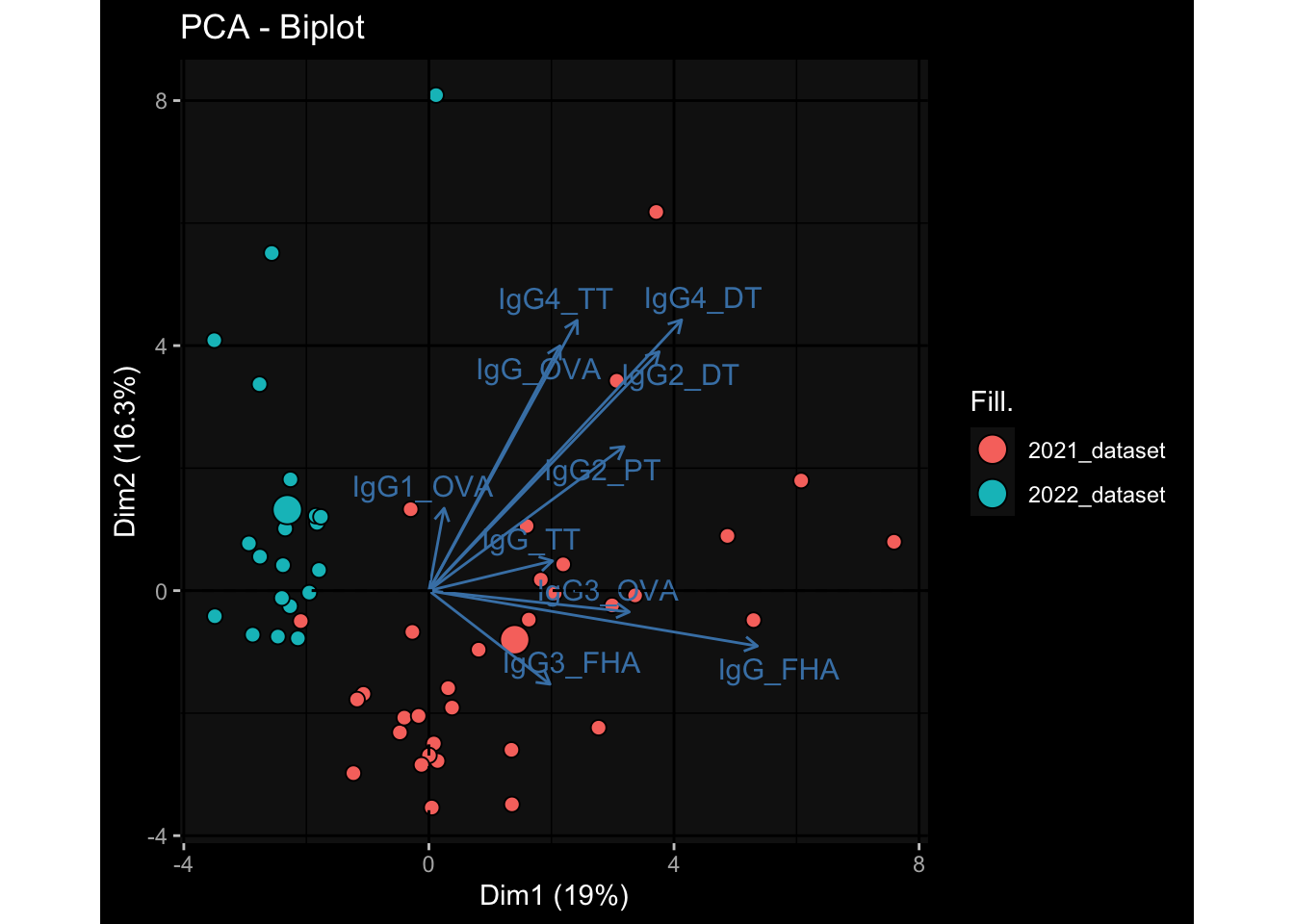
3.3 Plasma Antibody Levels
3.4 Plasma Cytokine Concentration by Legendplex
3.5 Plasma Cytokine Concentration by Olink
No further processing needed I guess.
3.6 T Cell Activation
No further processing needed I guess.
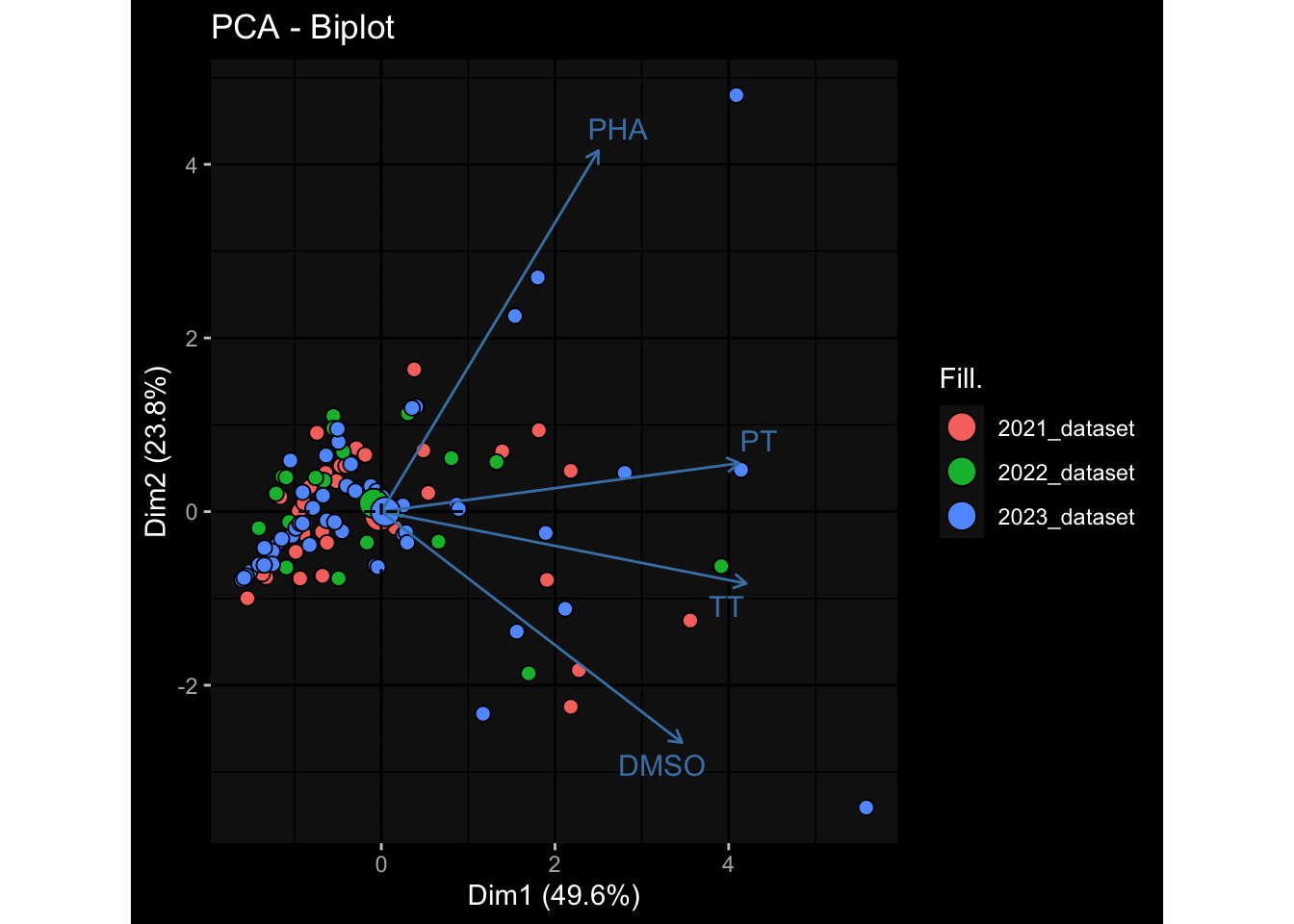
3.7 T Cell Polarization
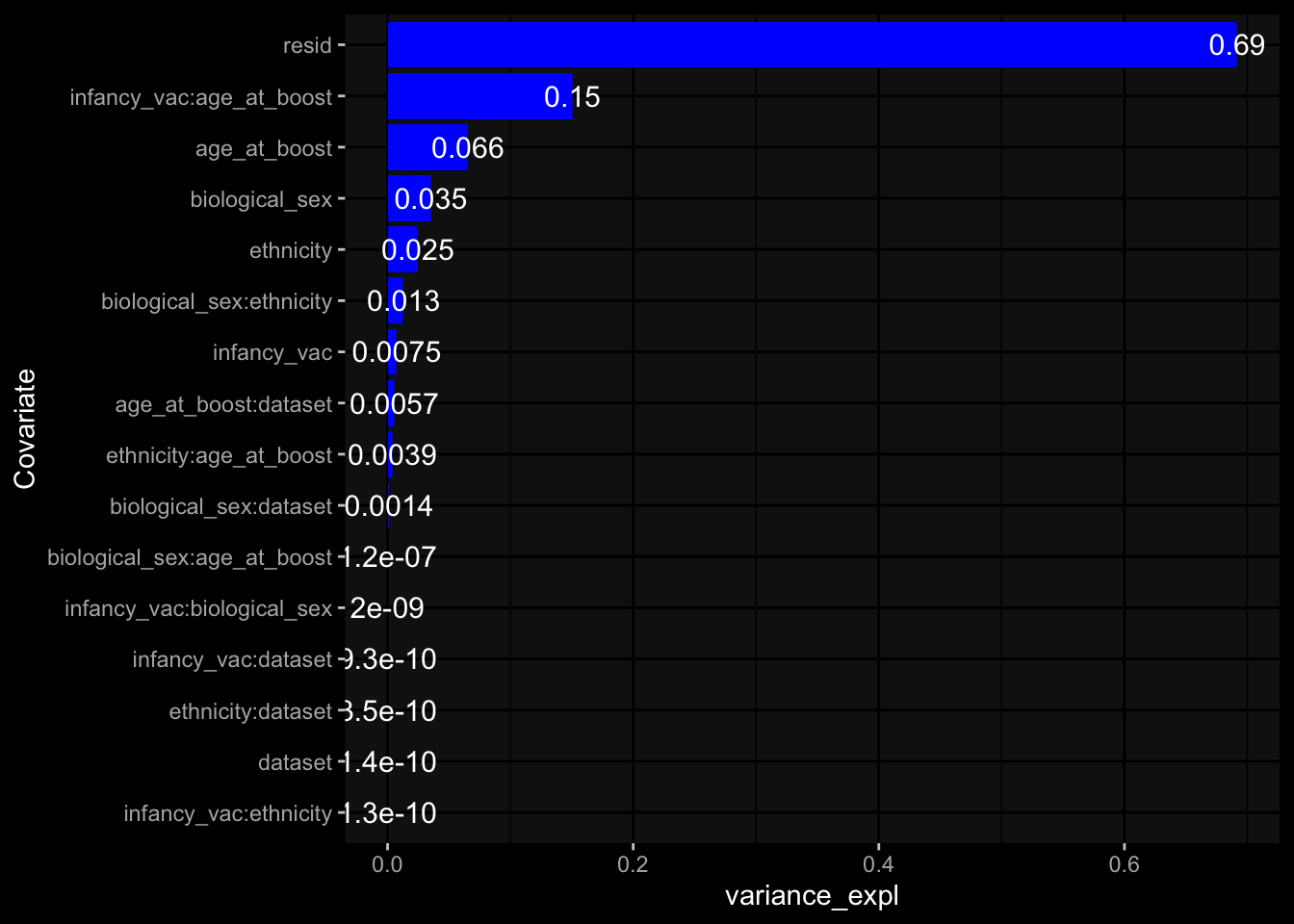
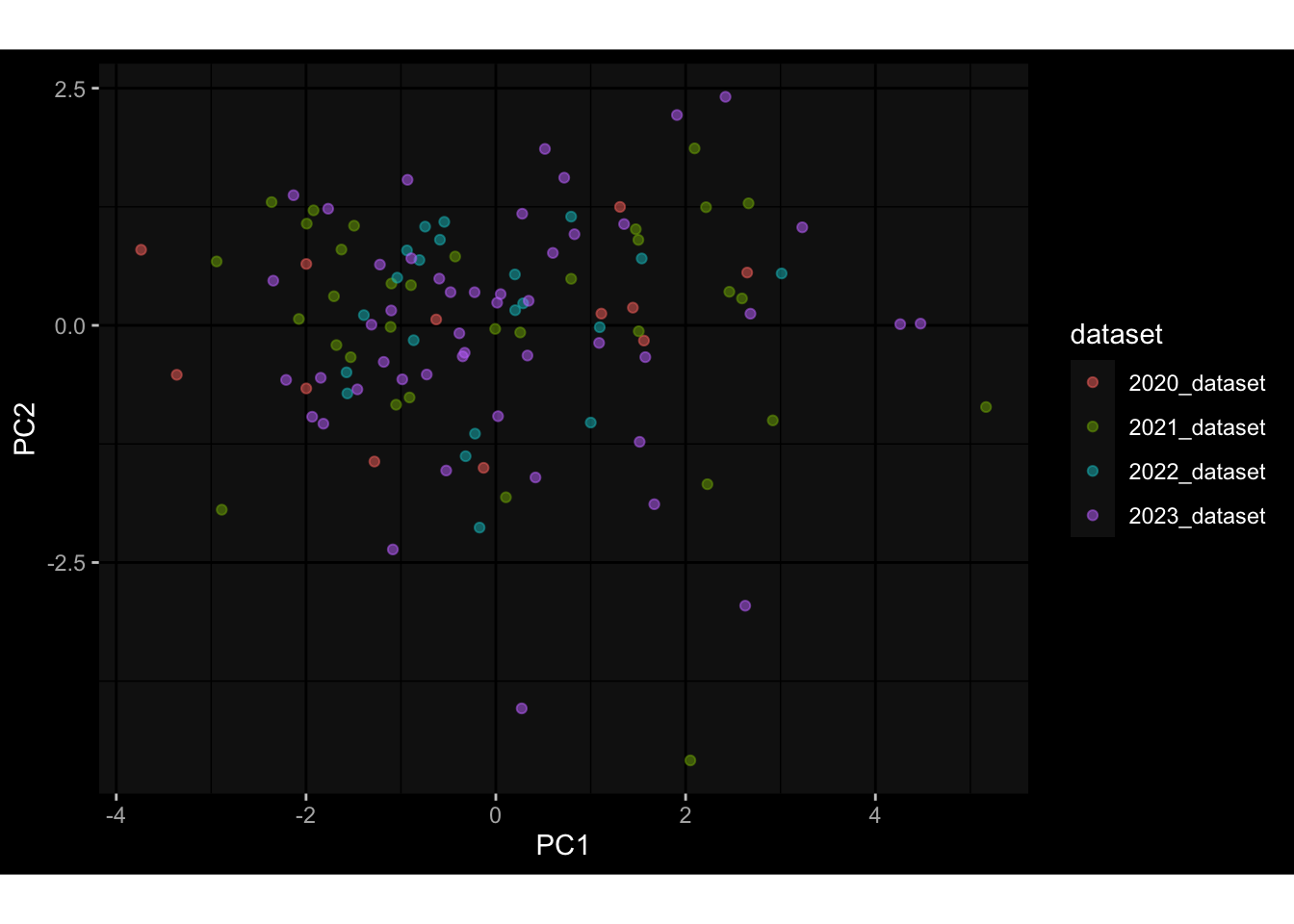
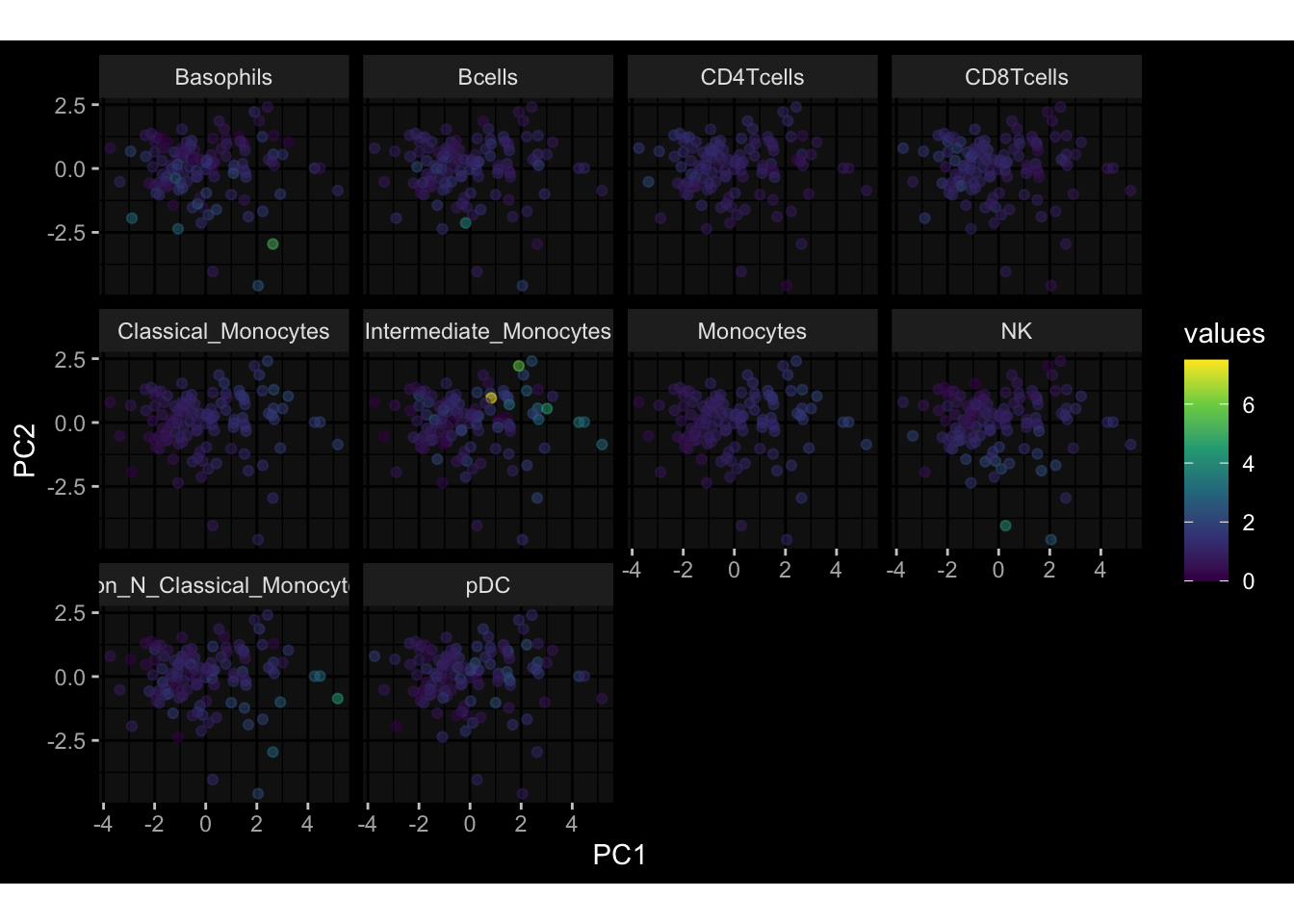
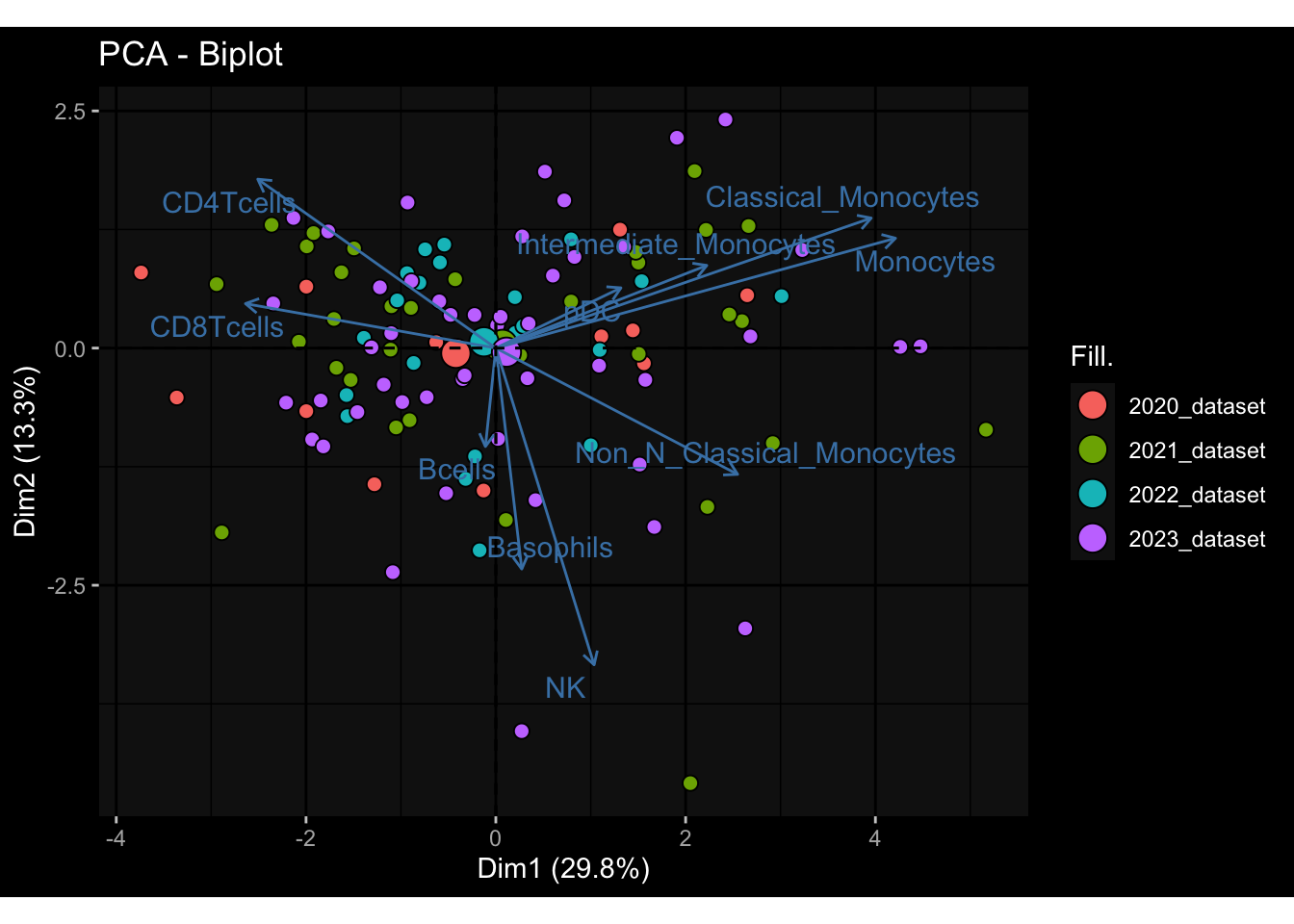
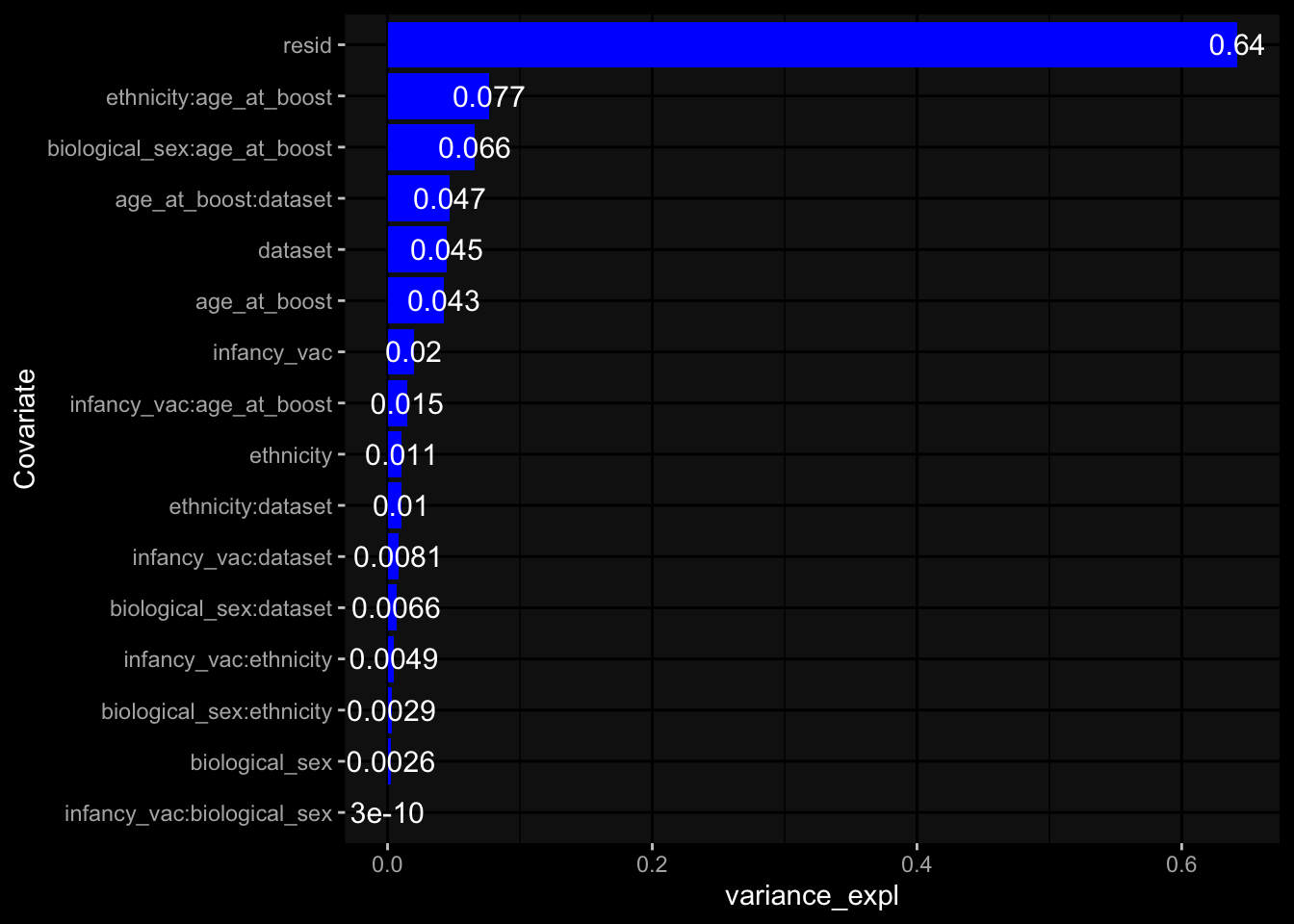
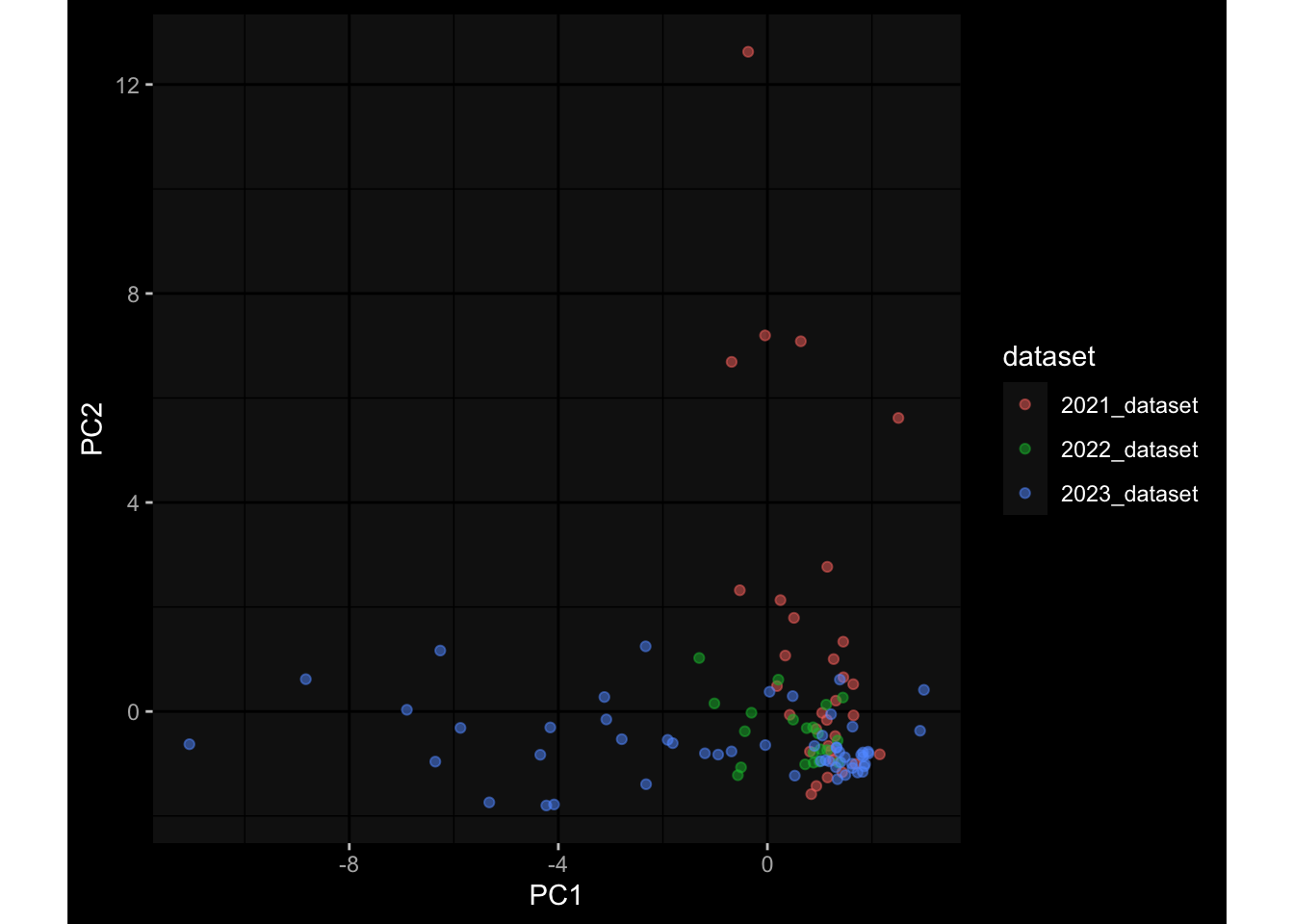
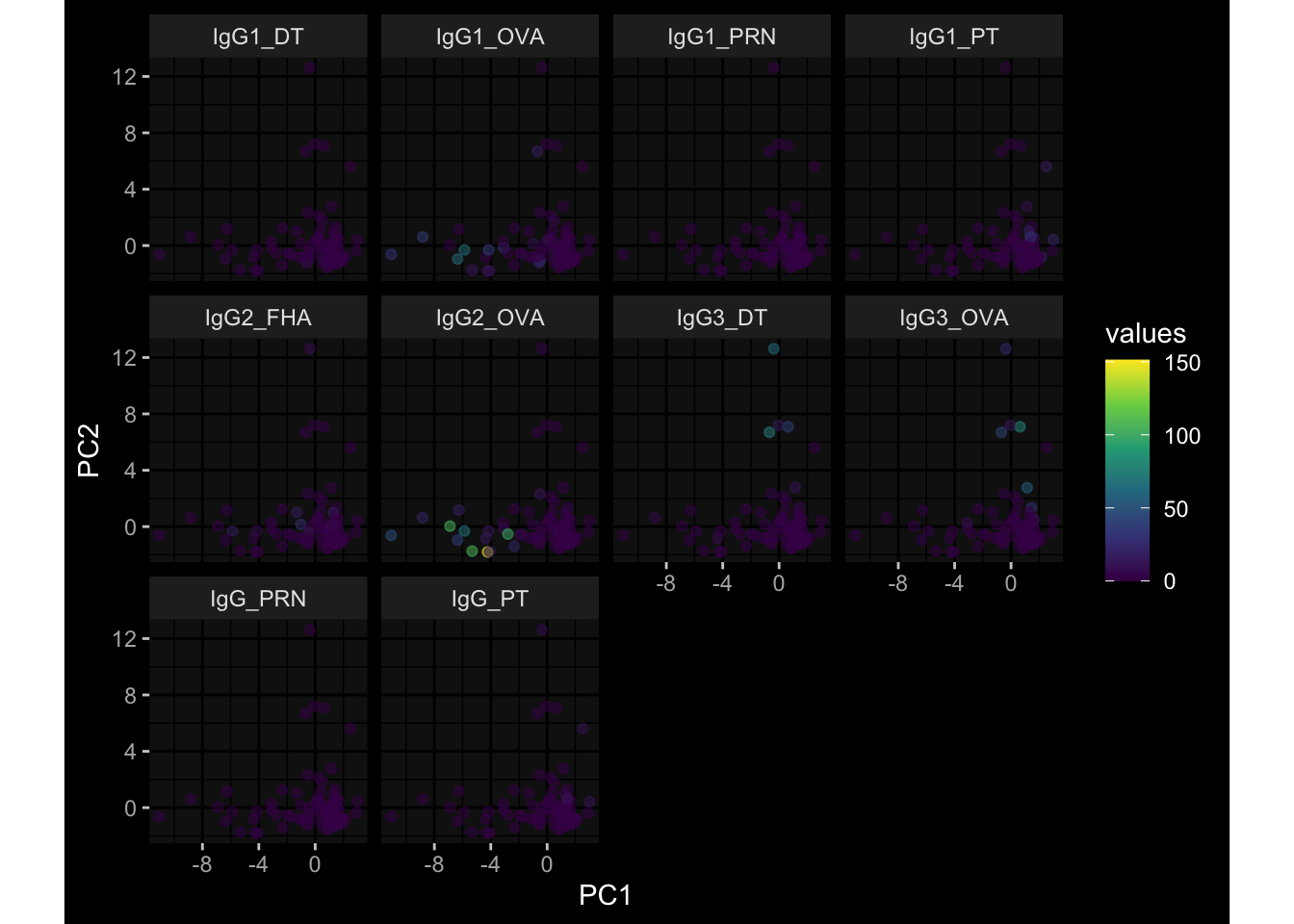
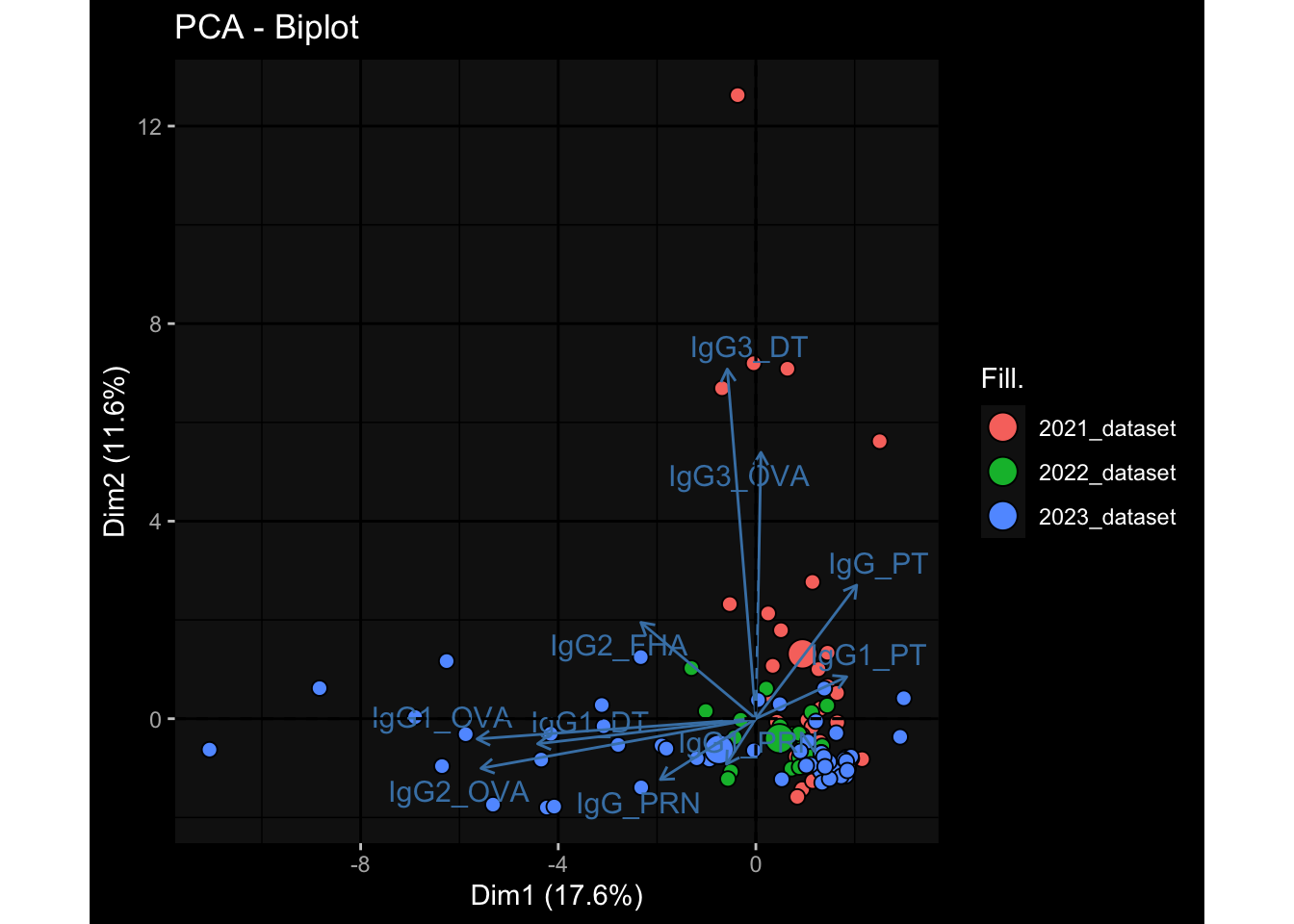
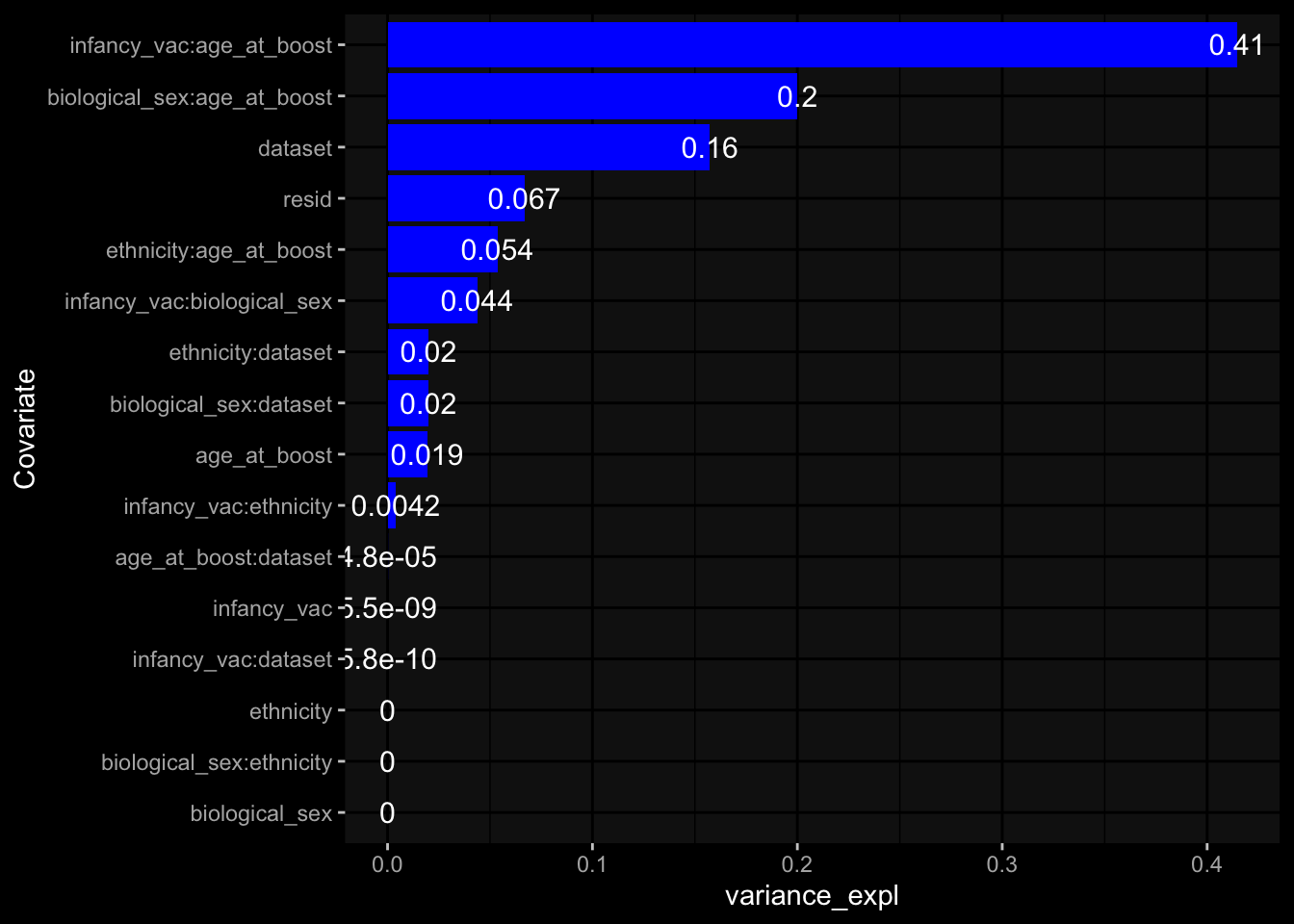
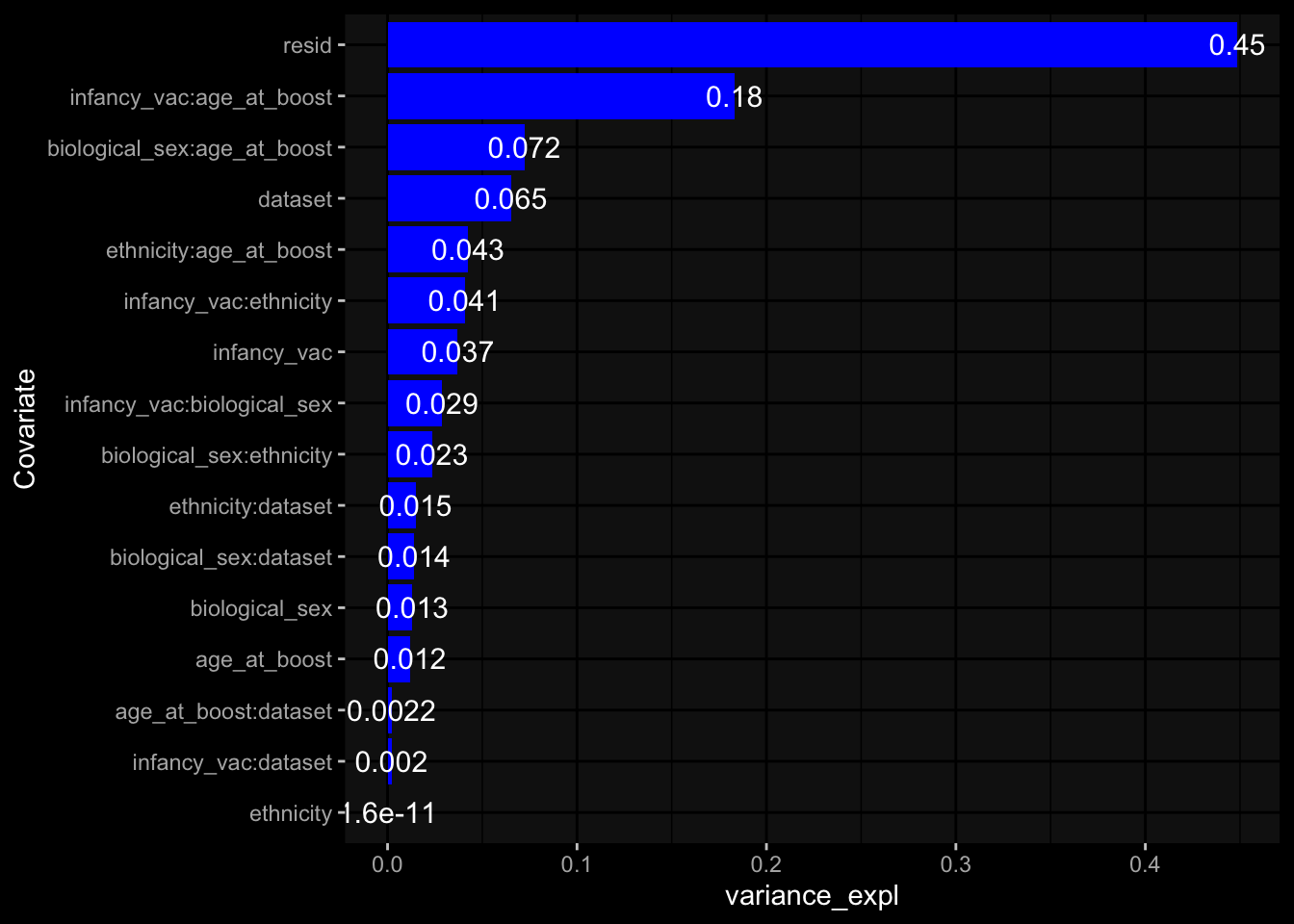
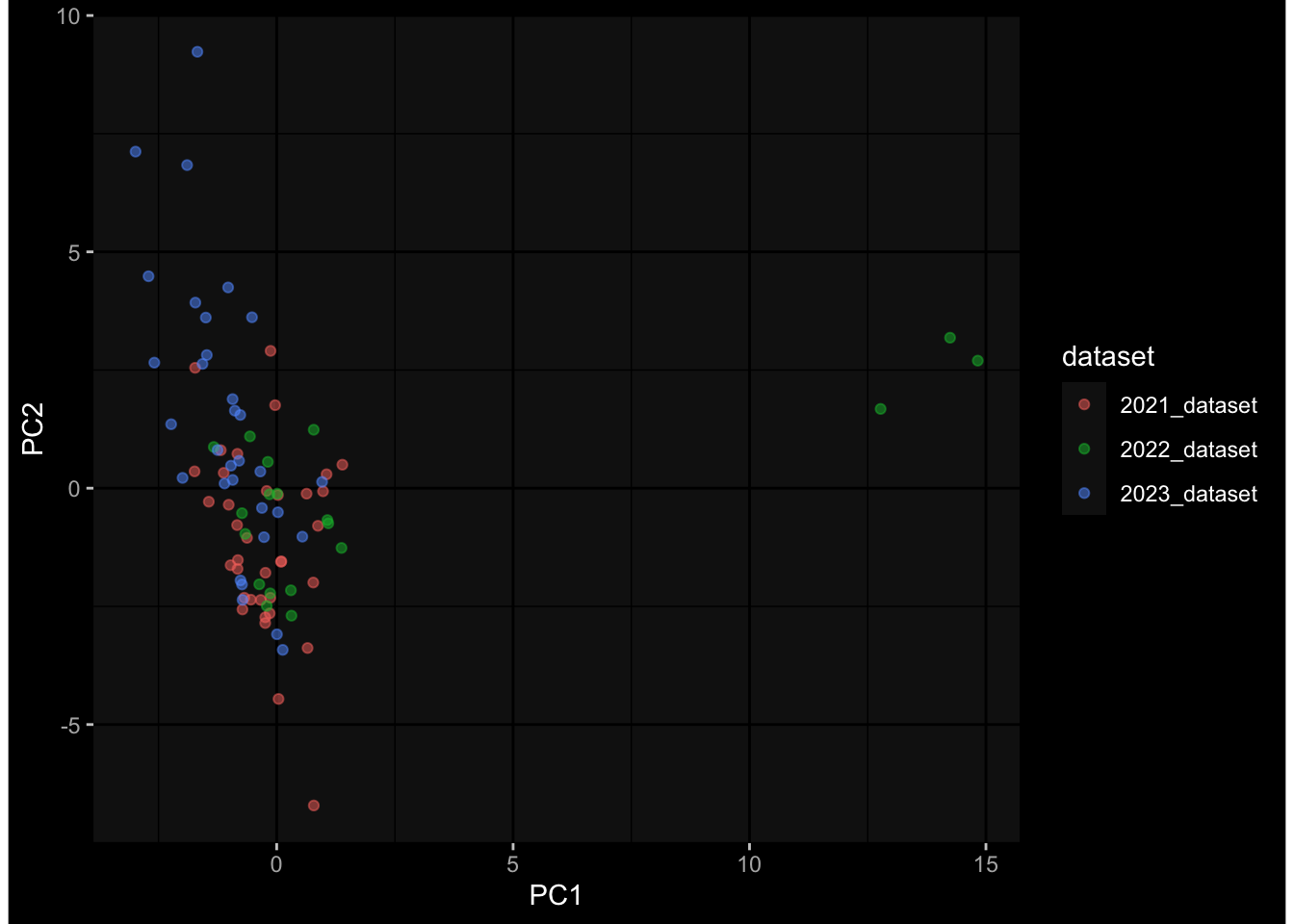
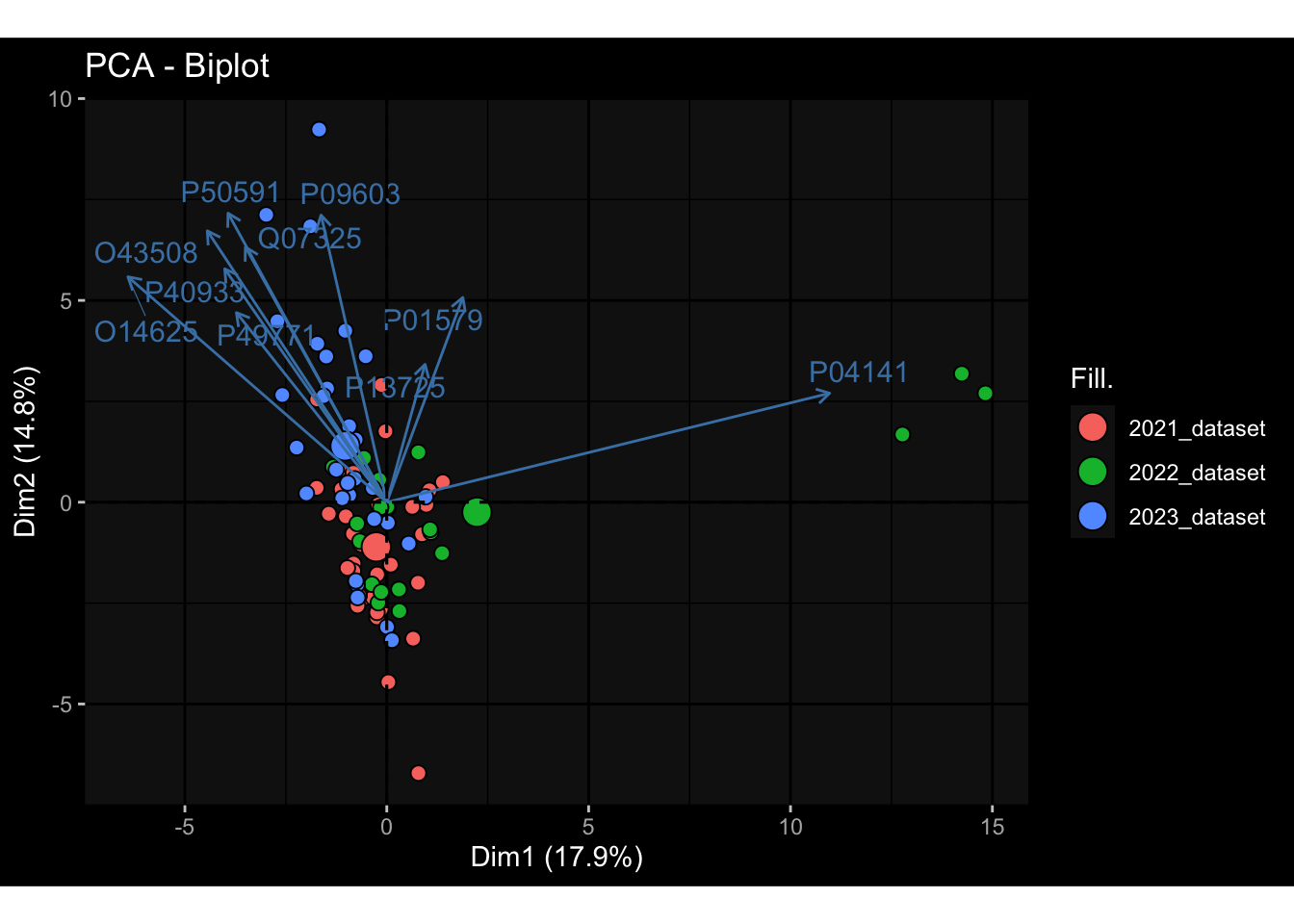
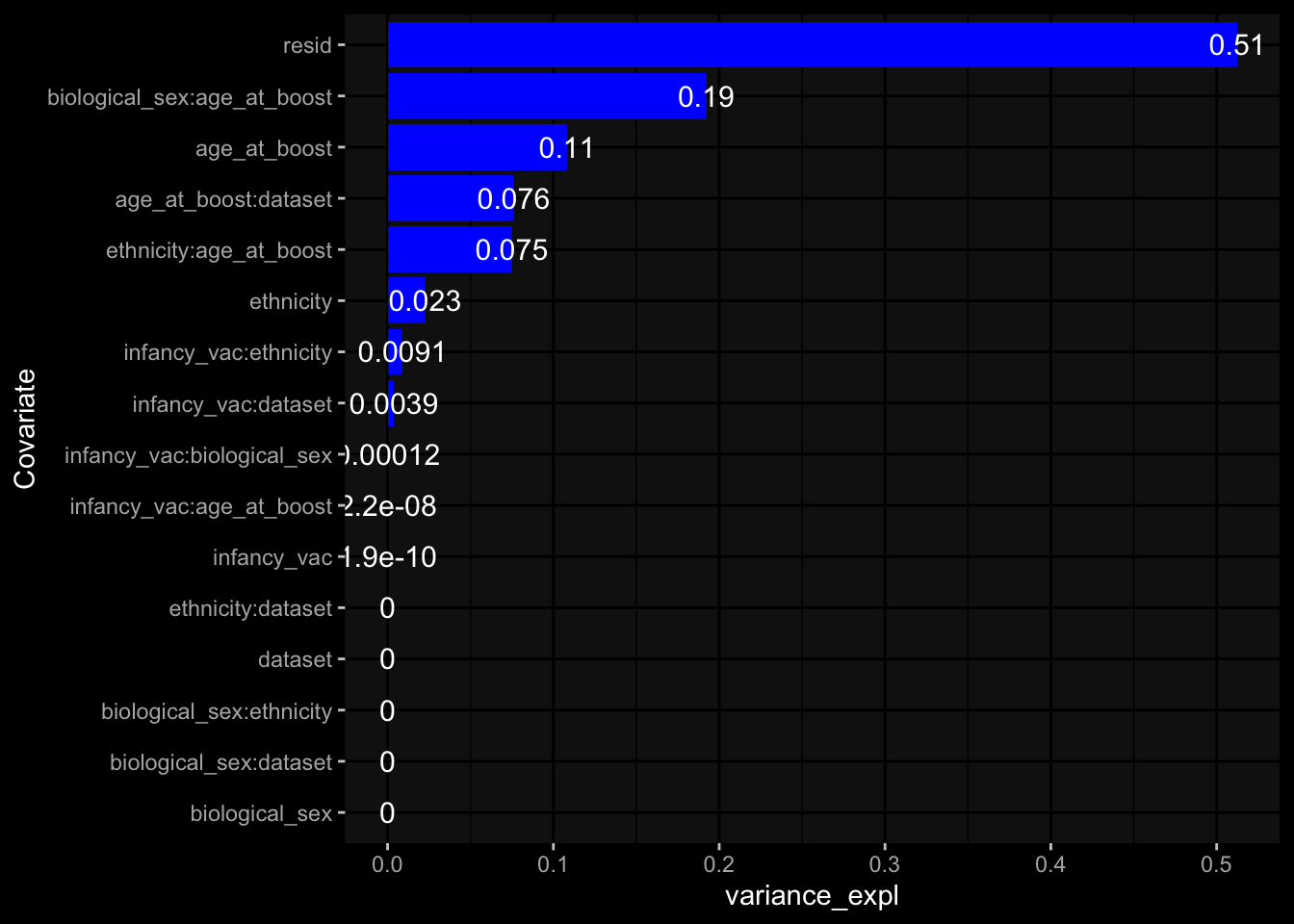
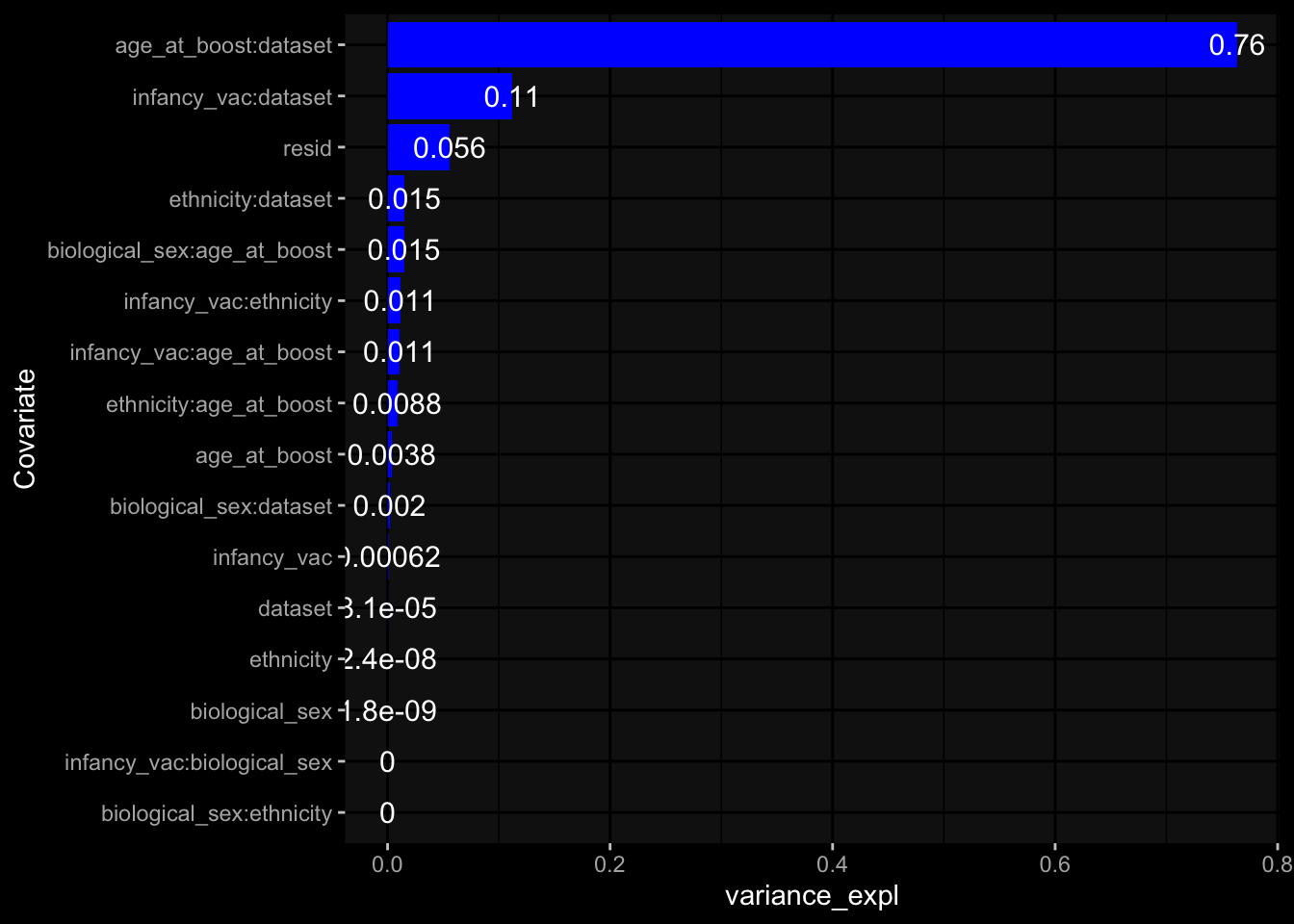
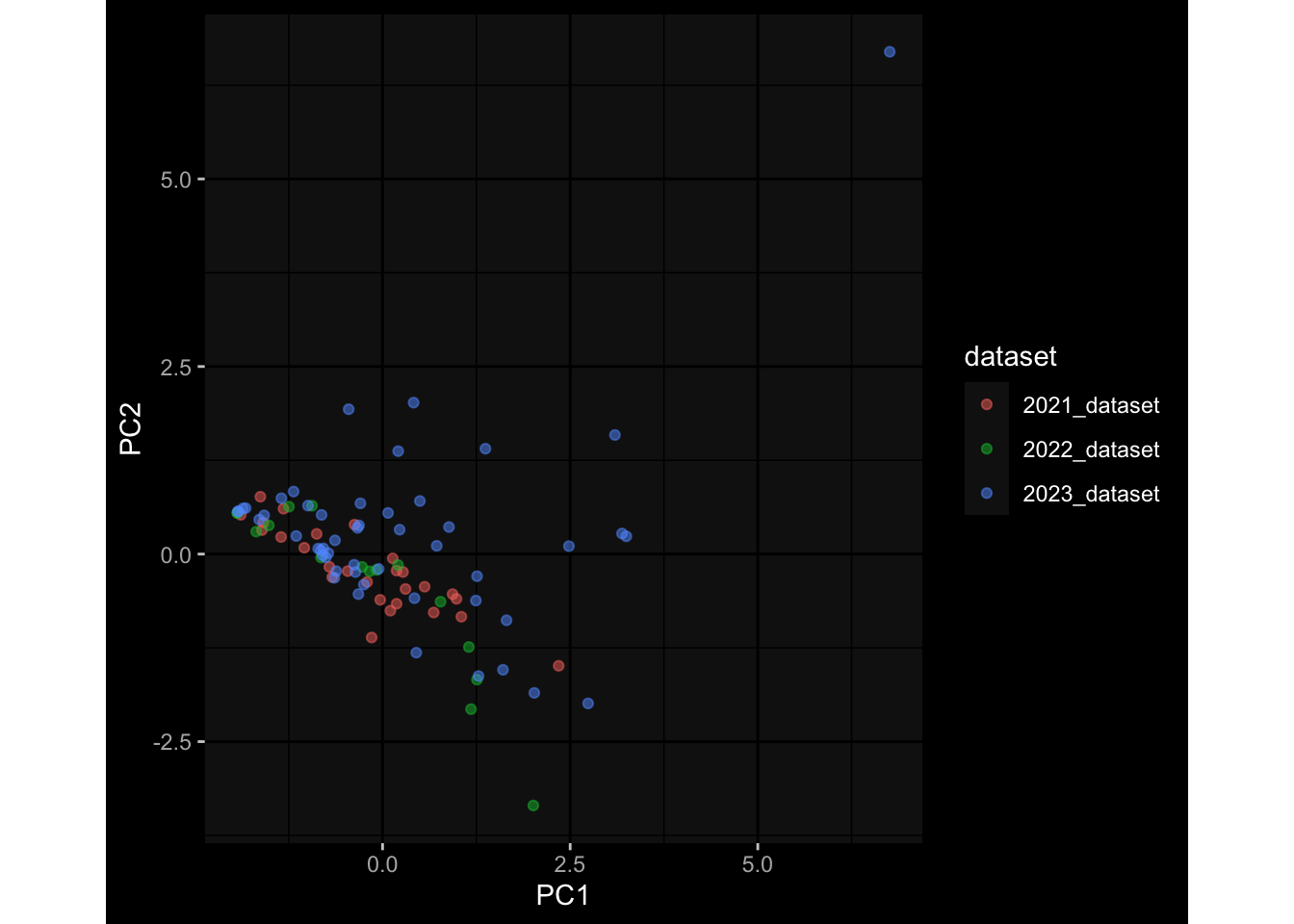
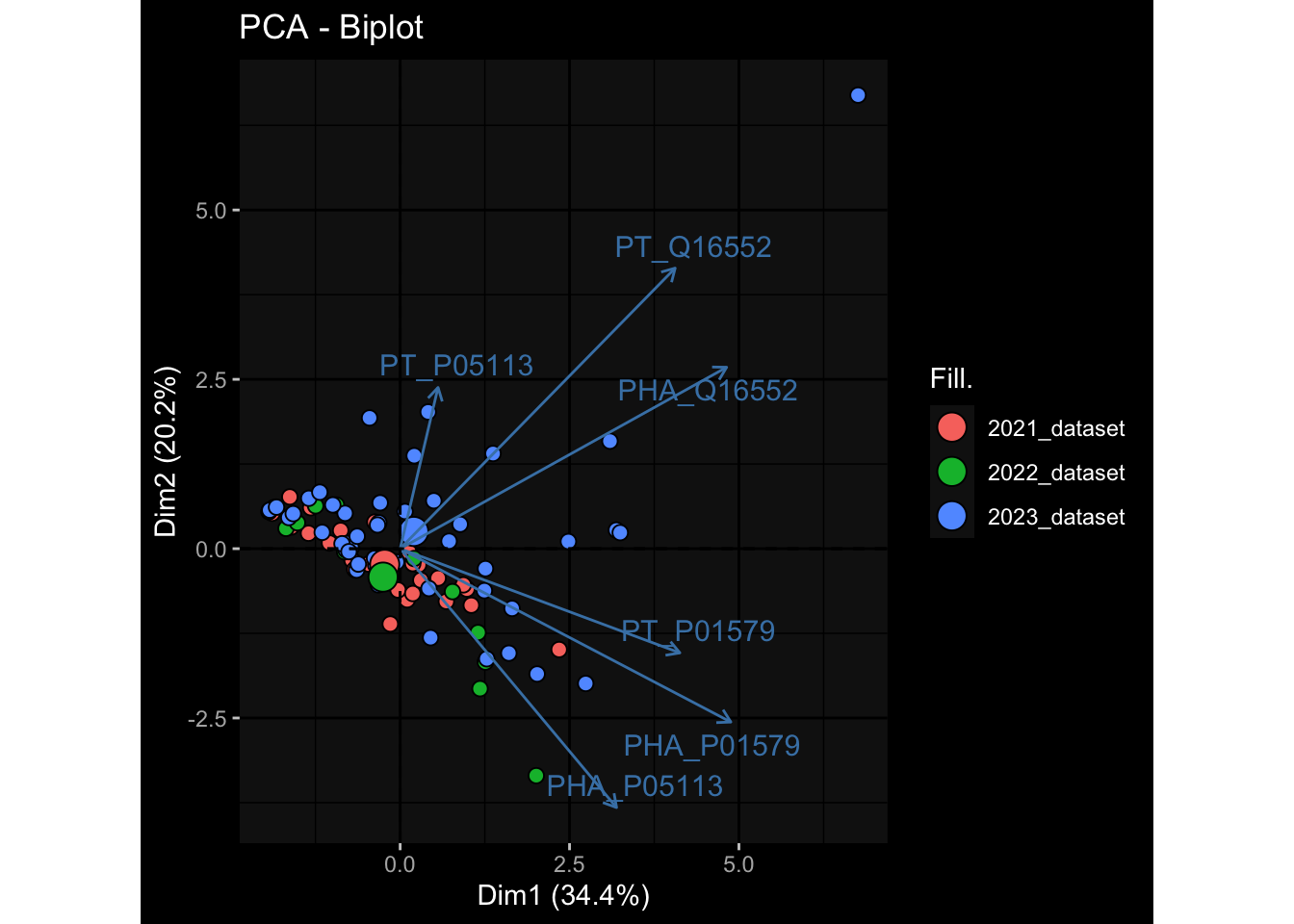
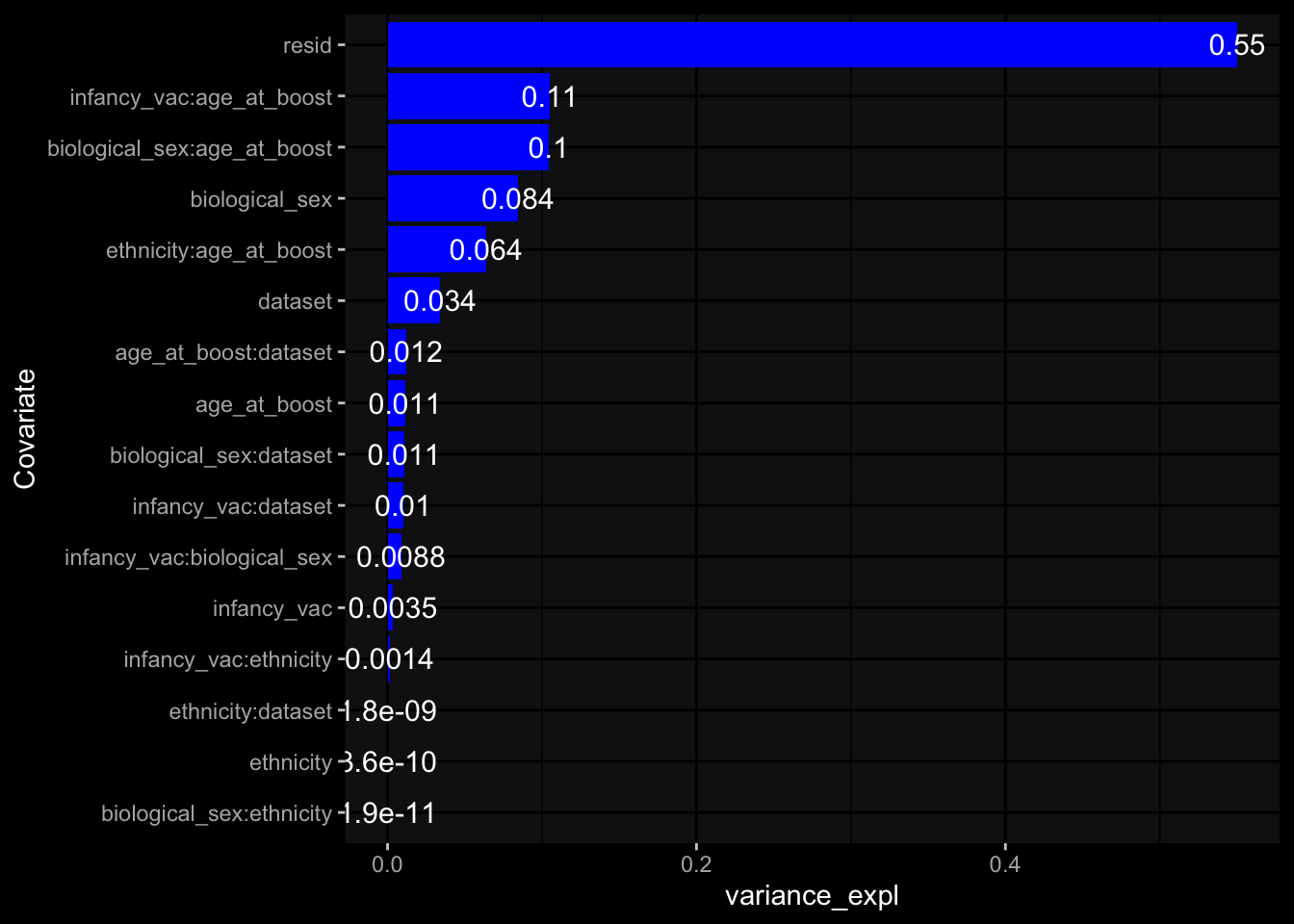
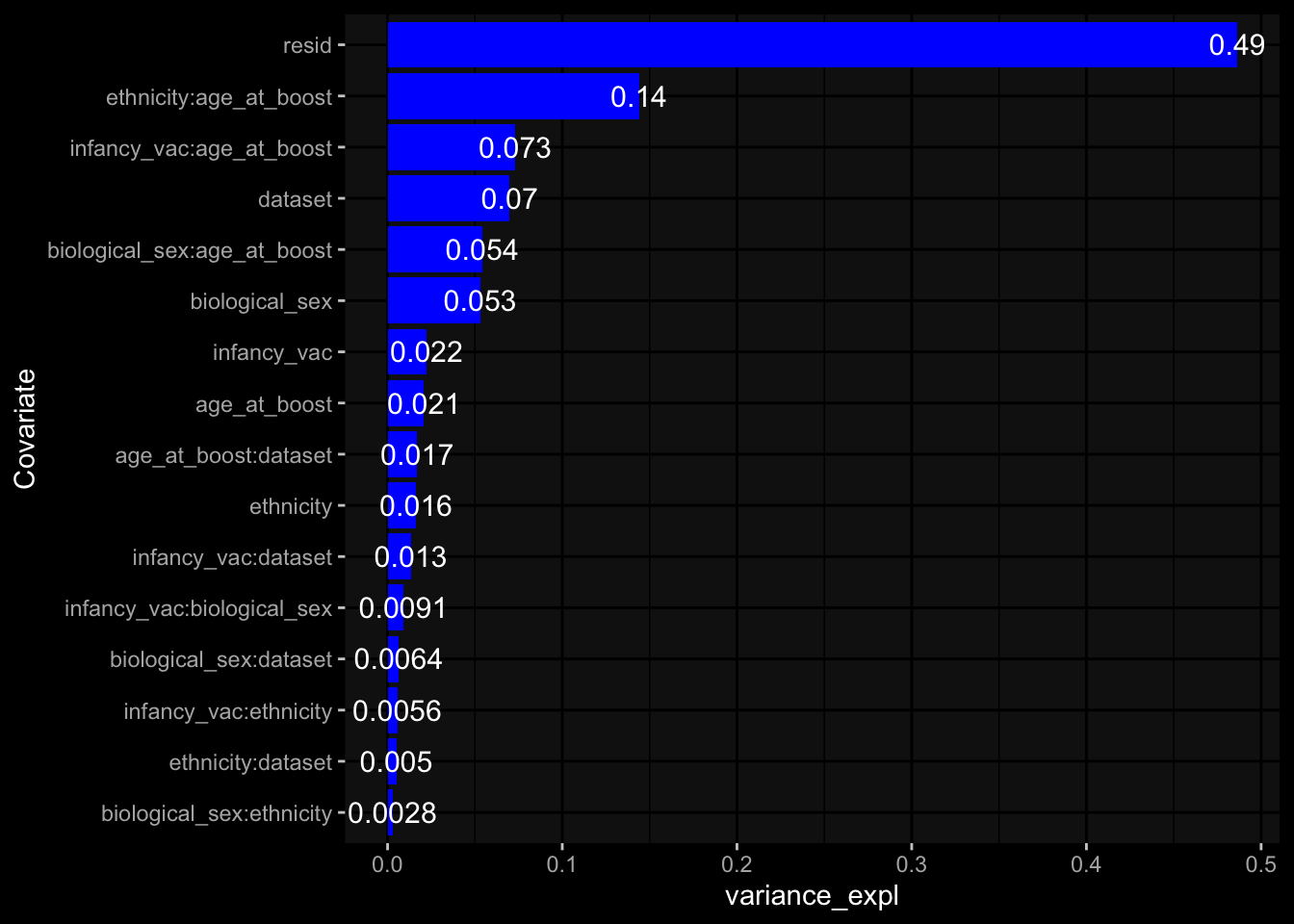
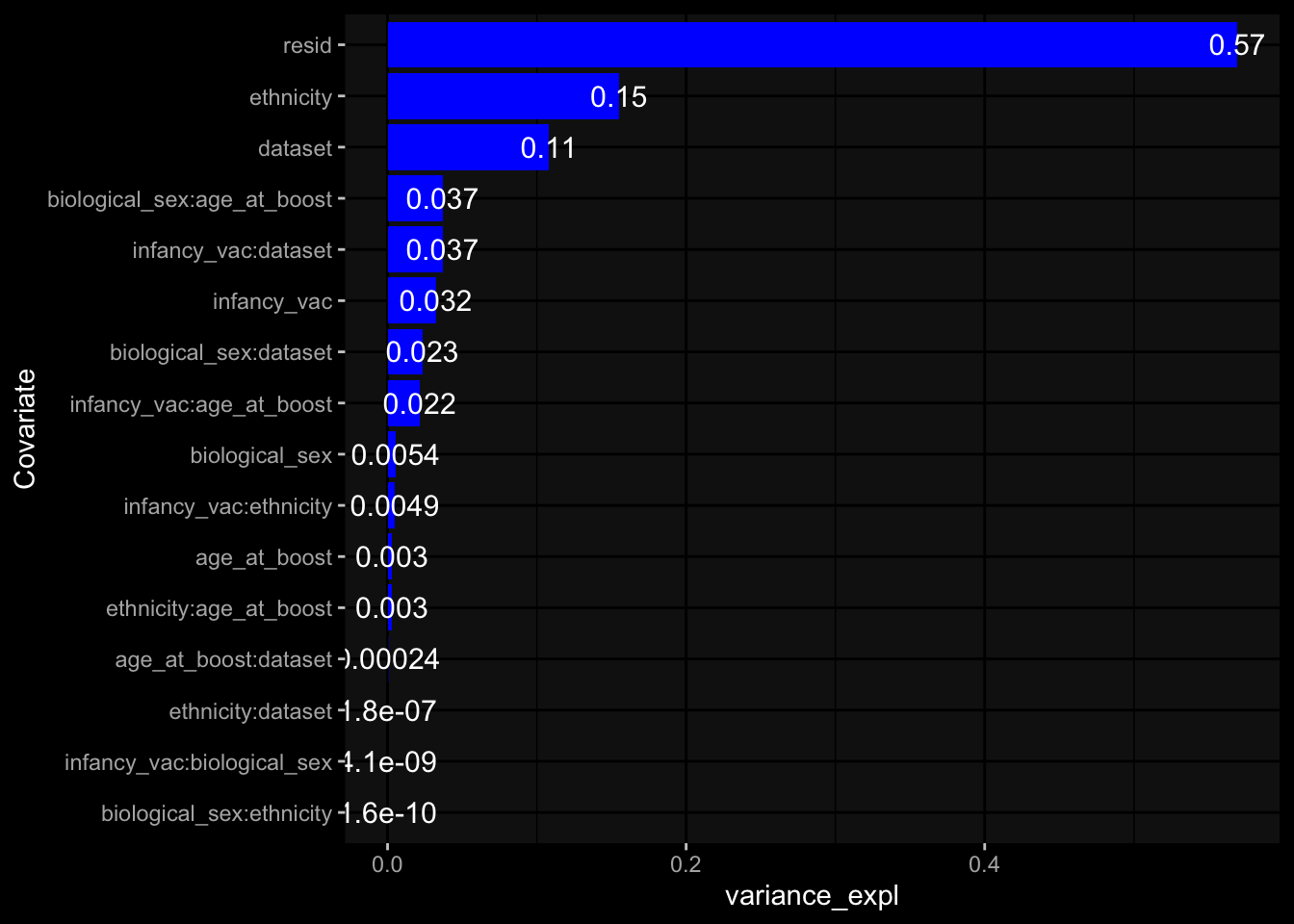
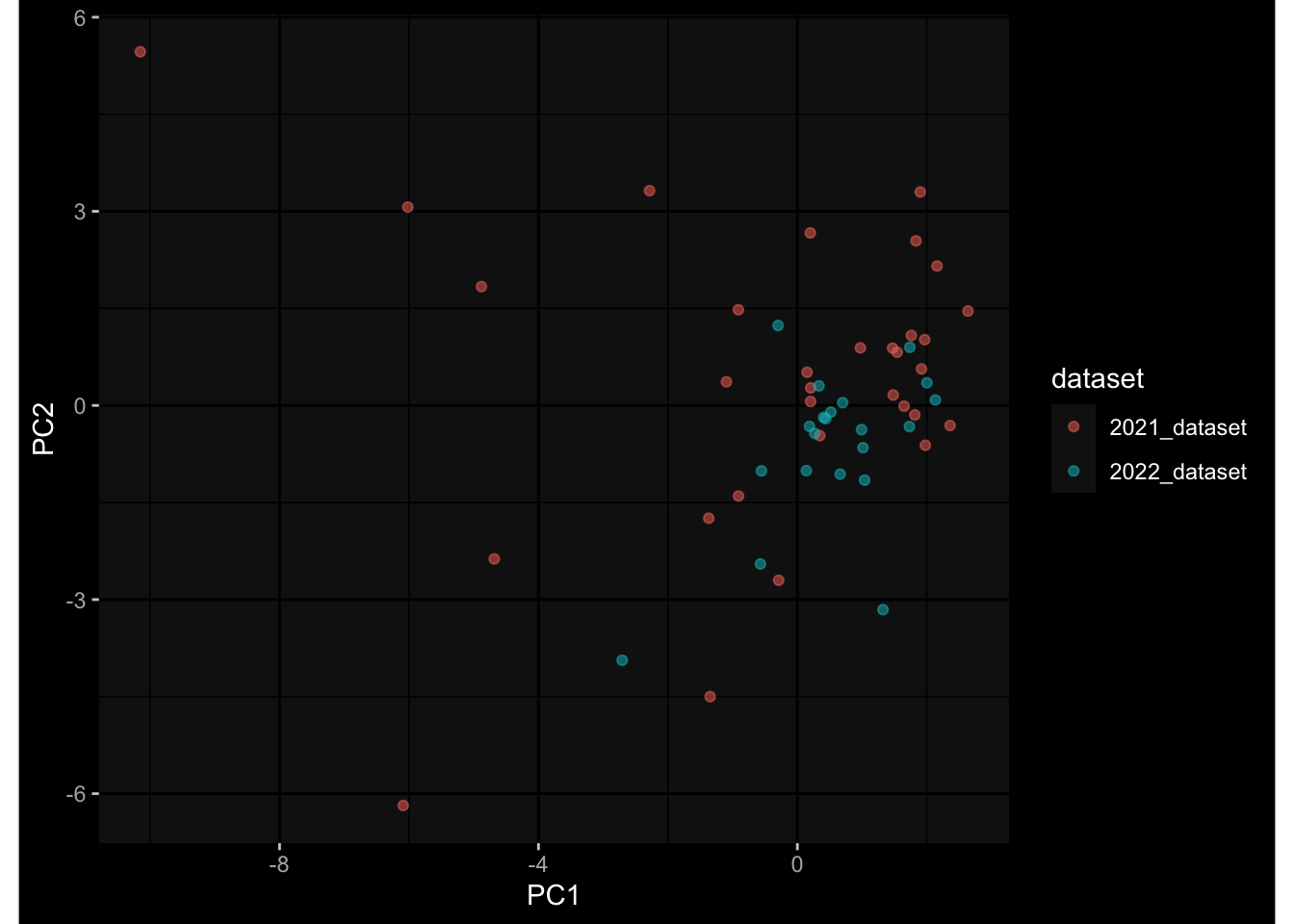
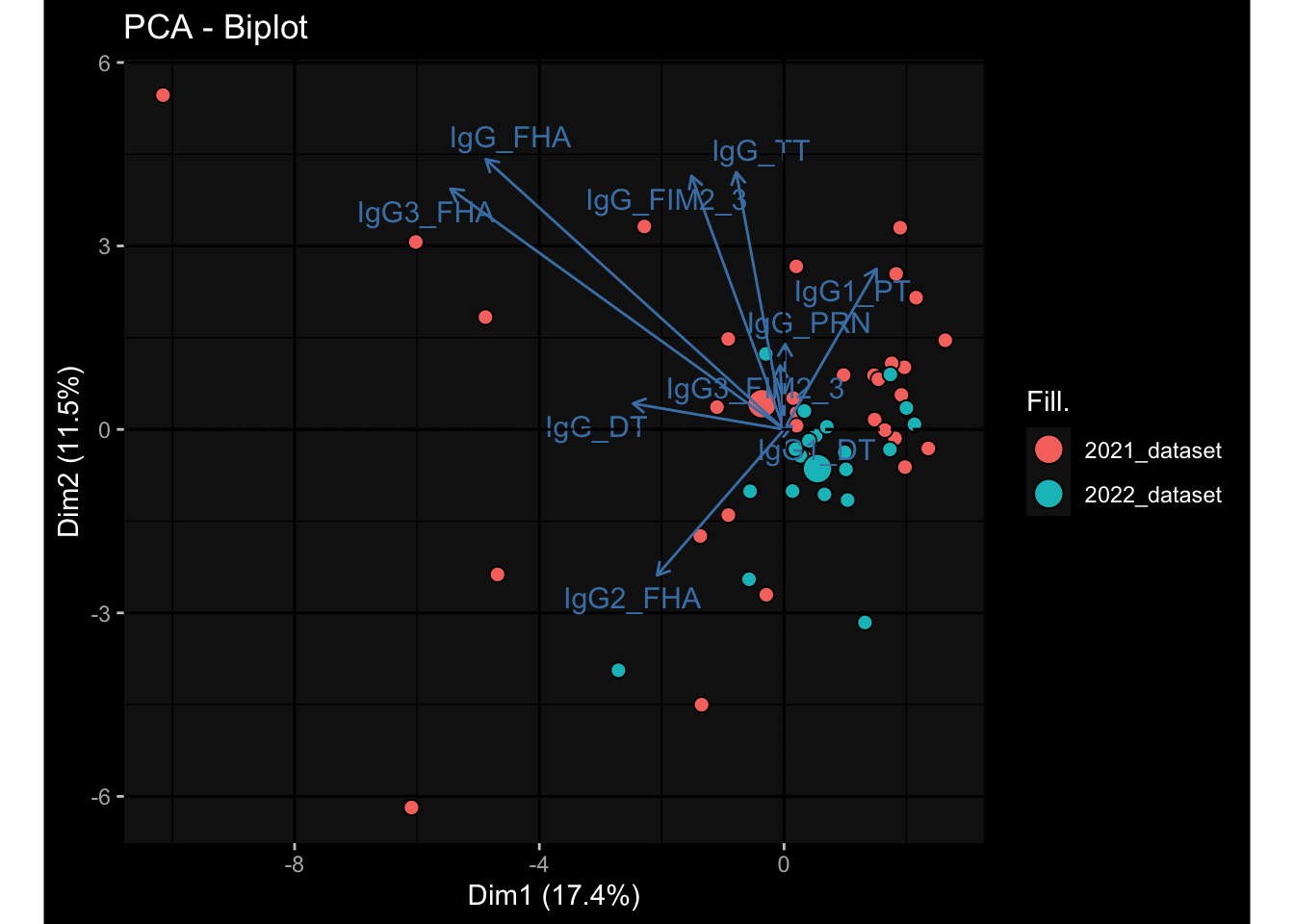
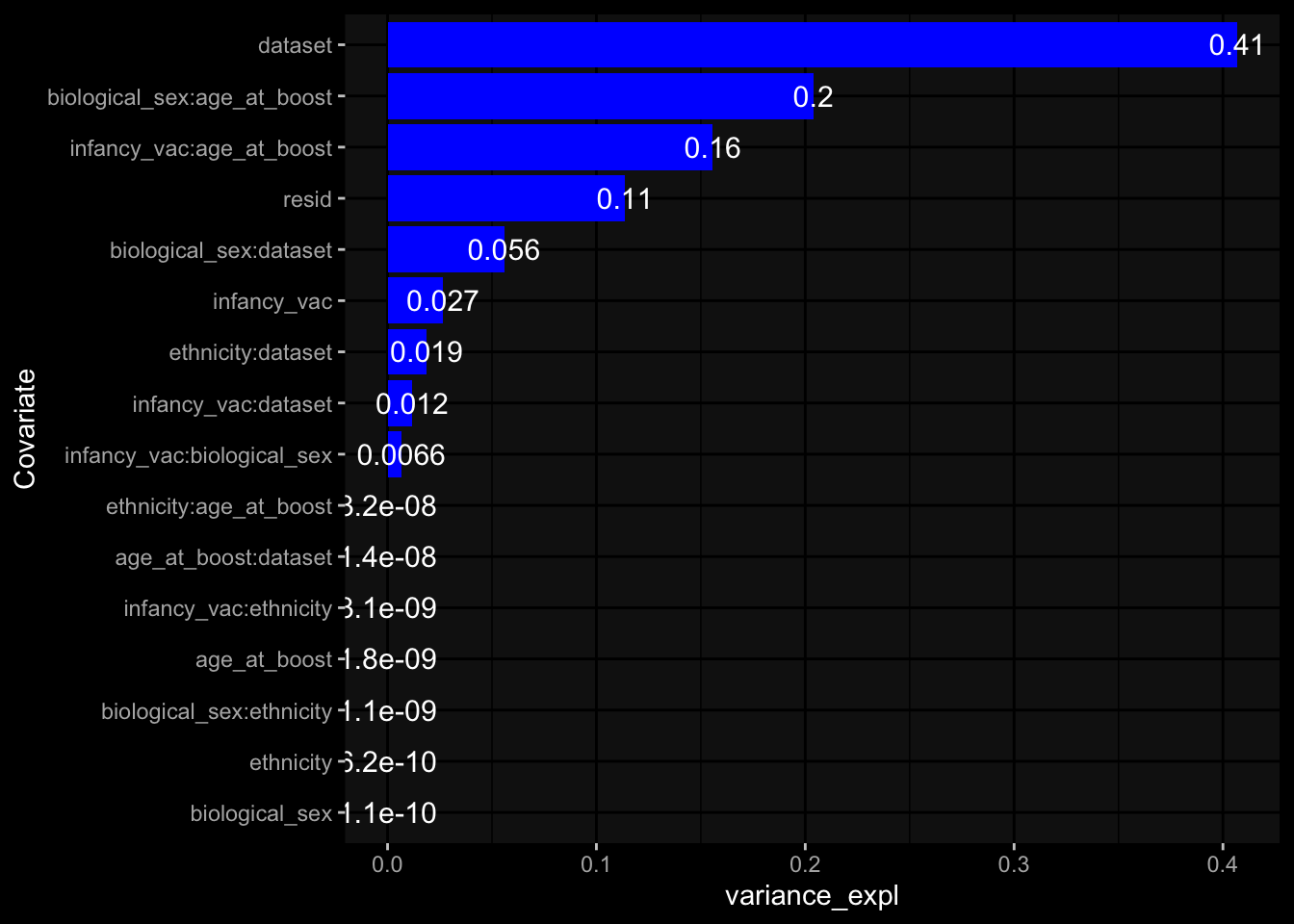
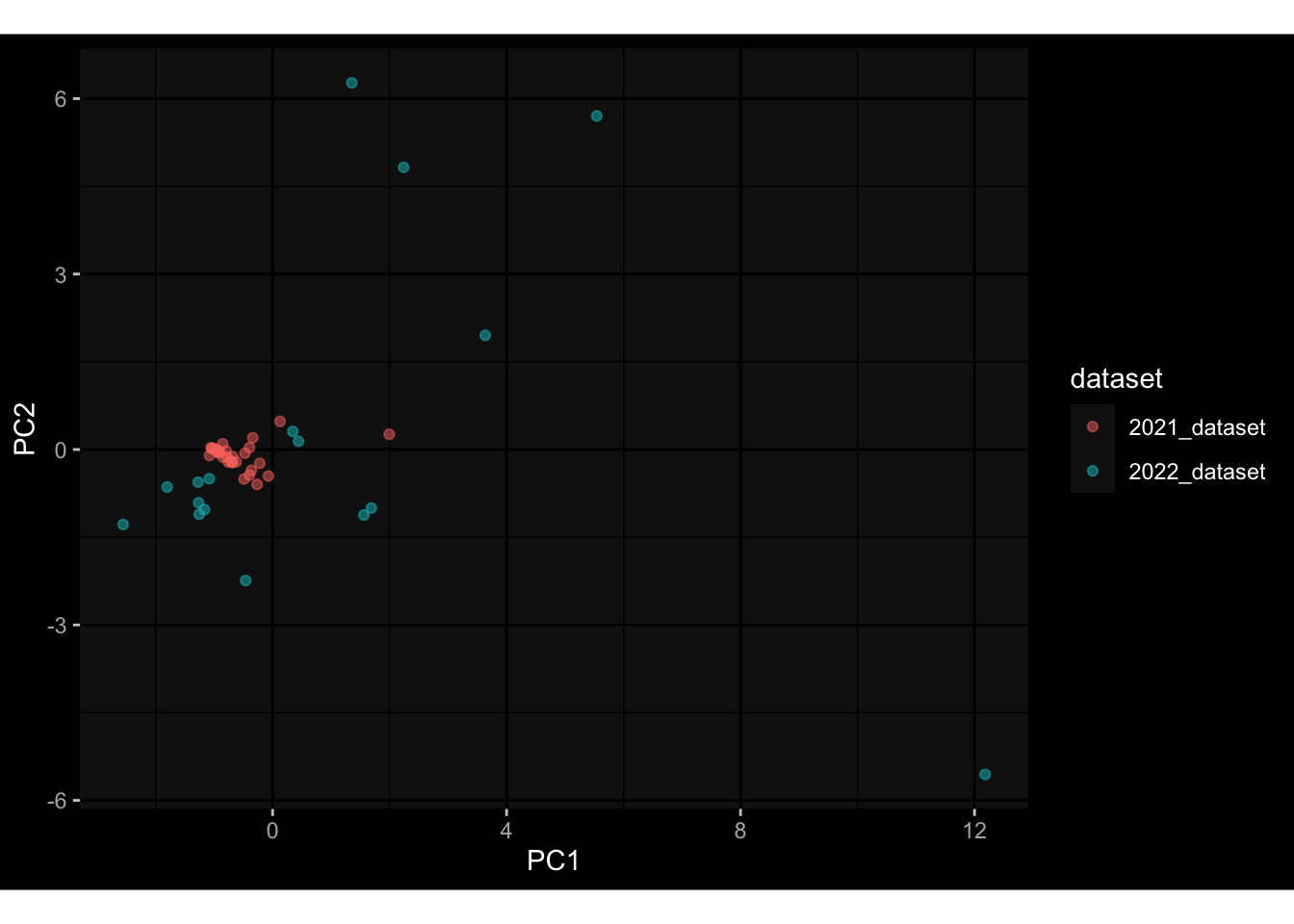
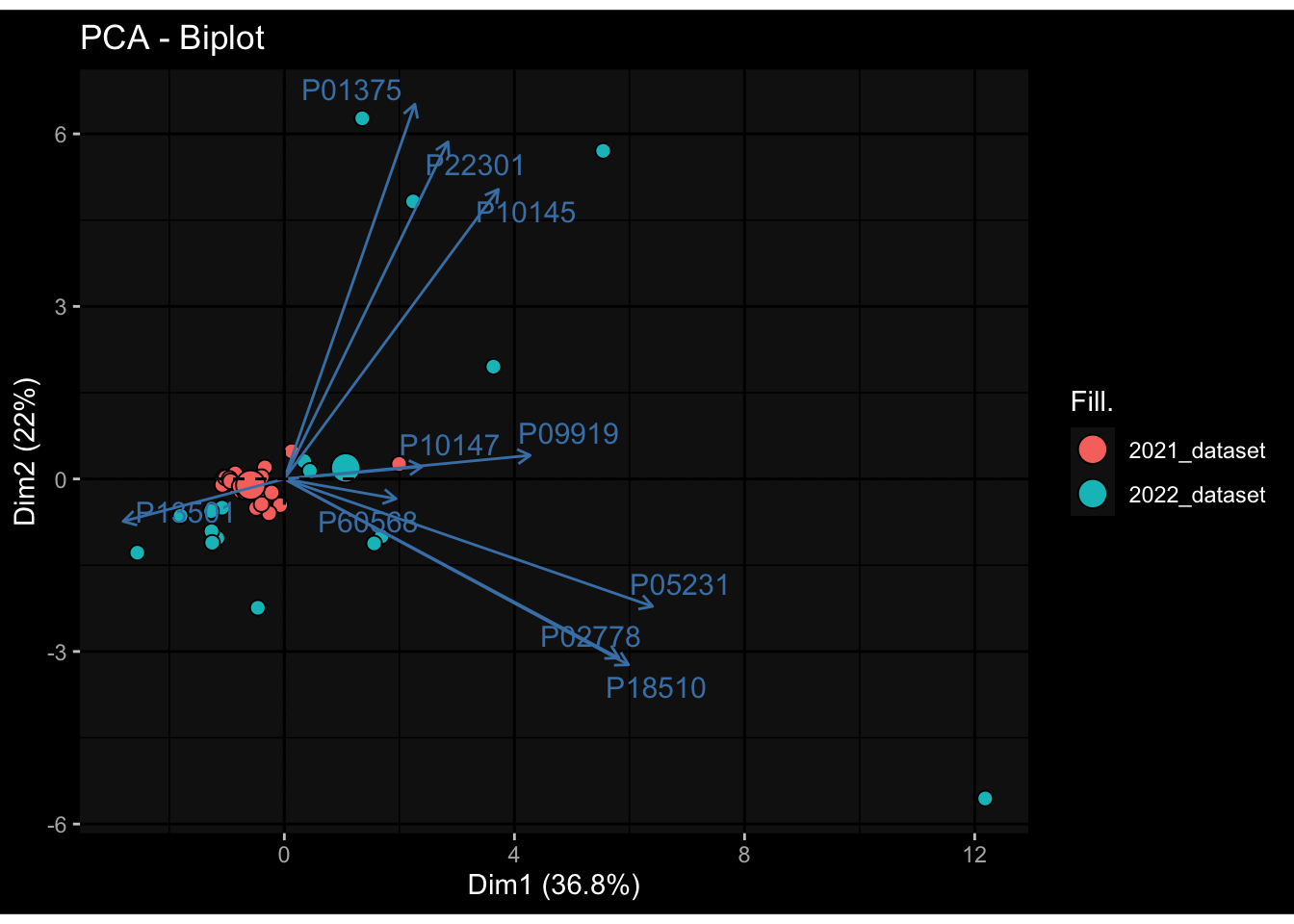
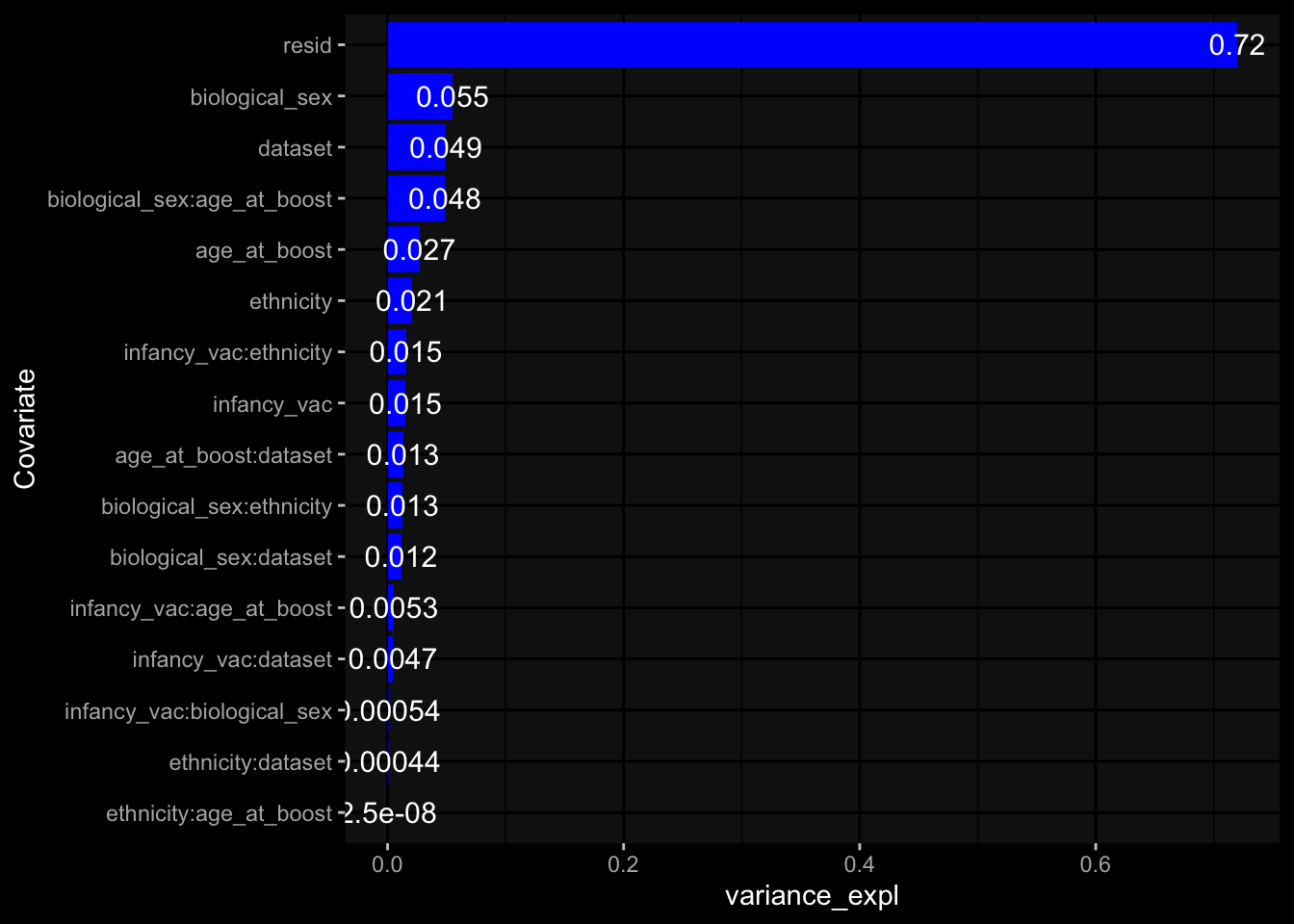
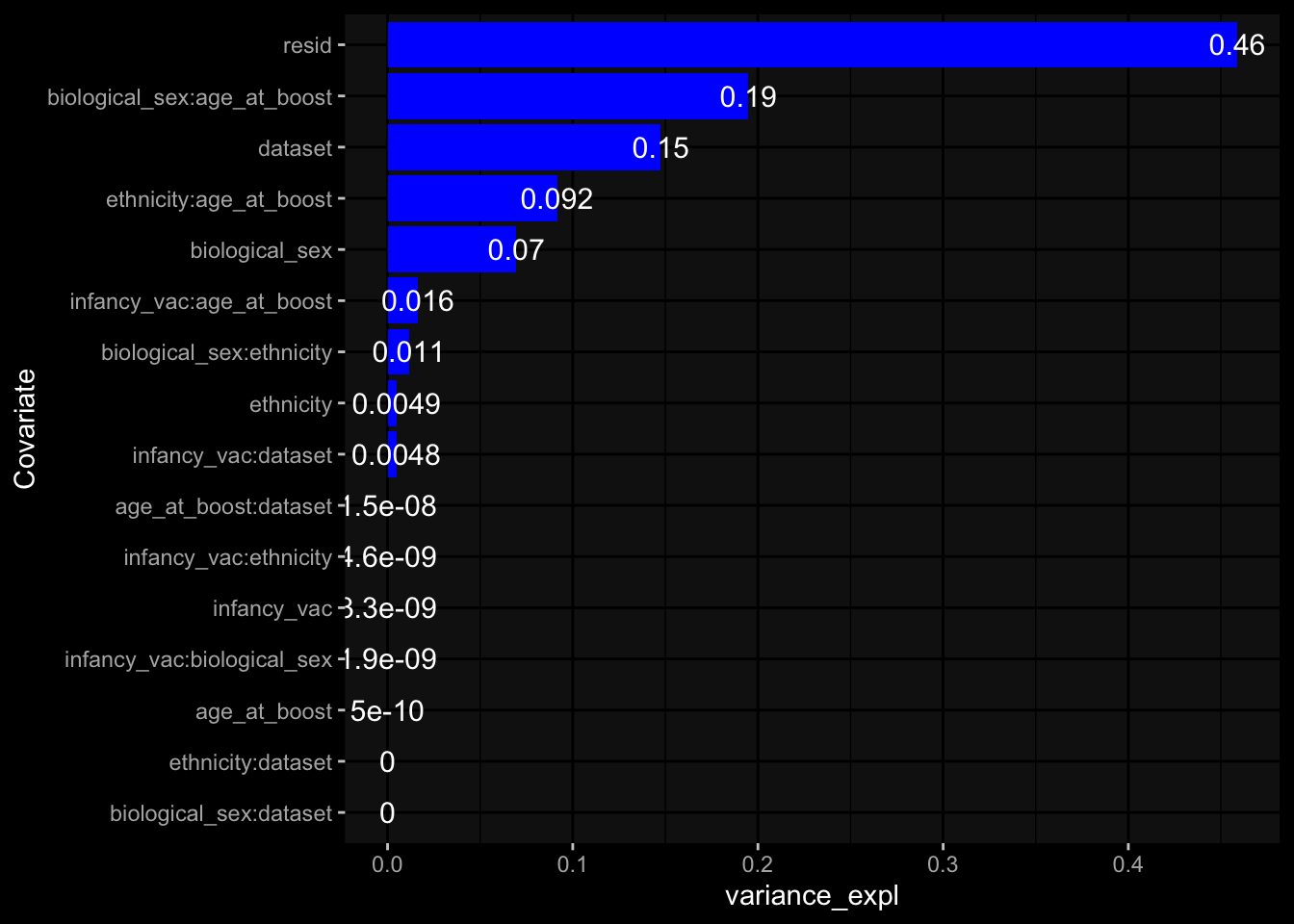
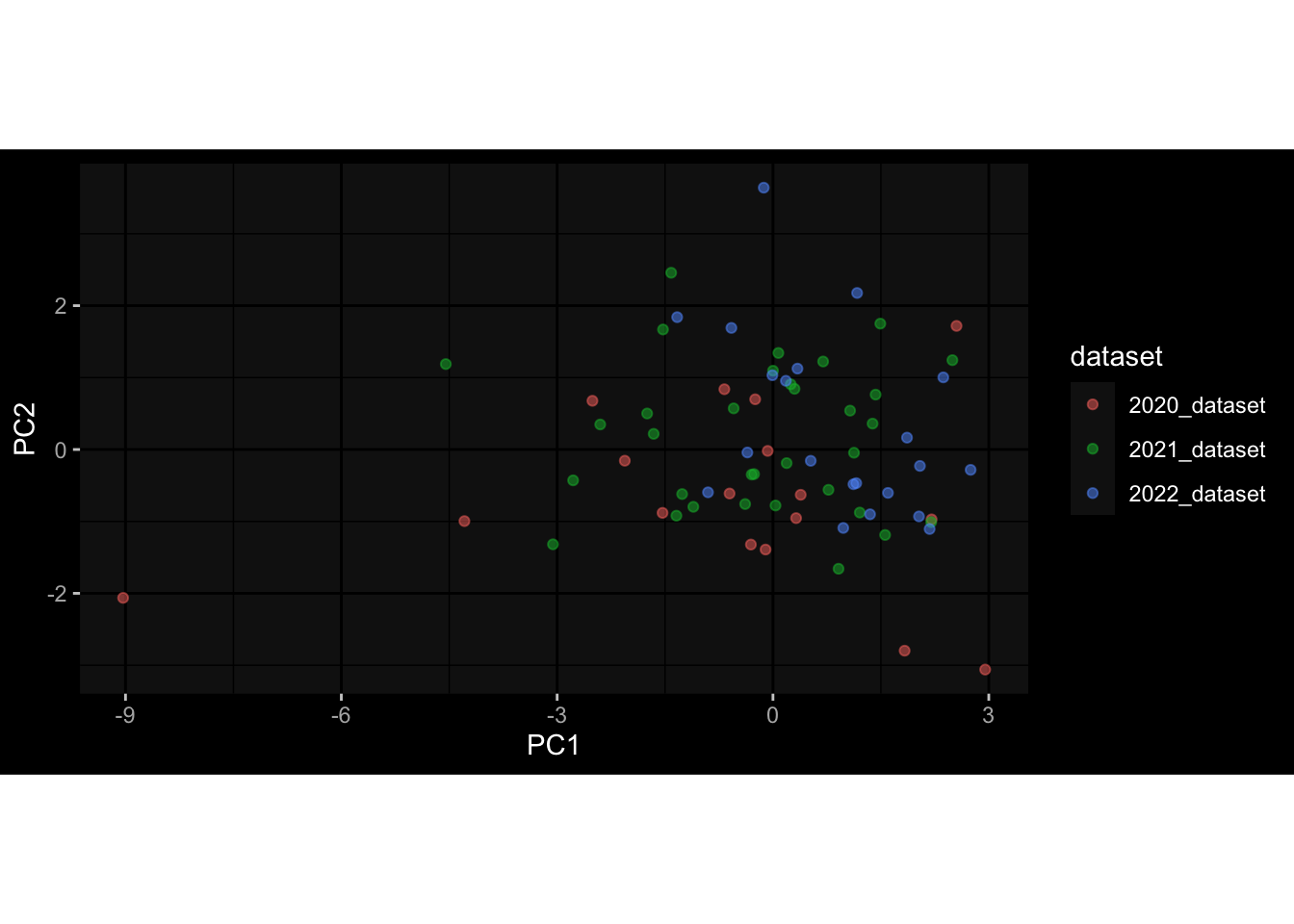
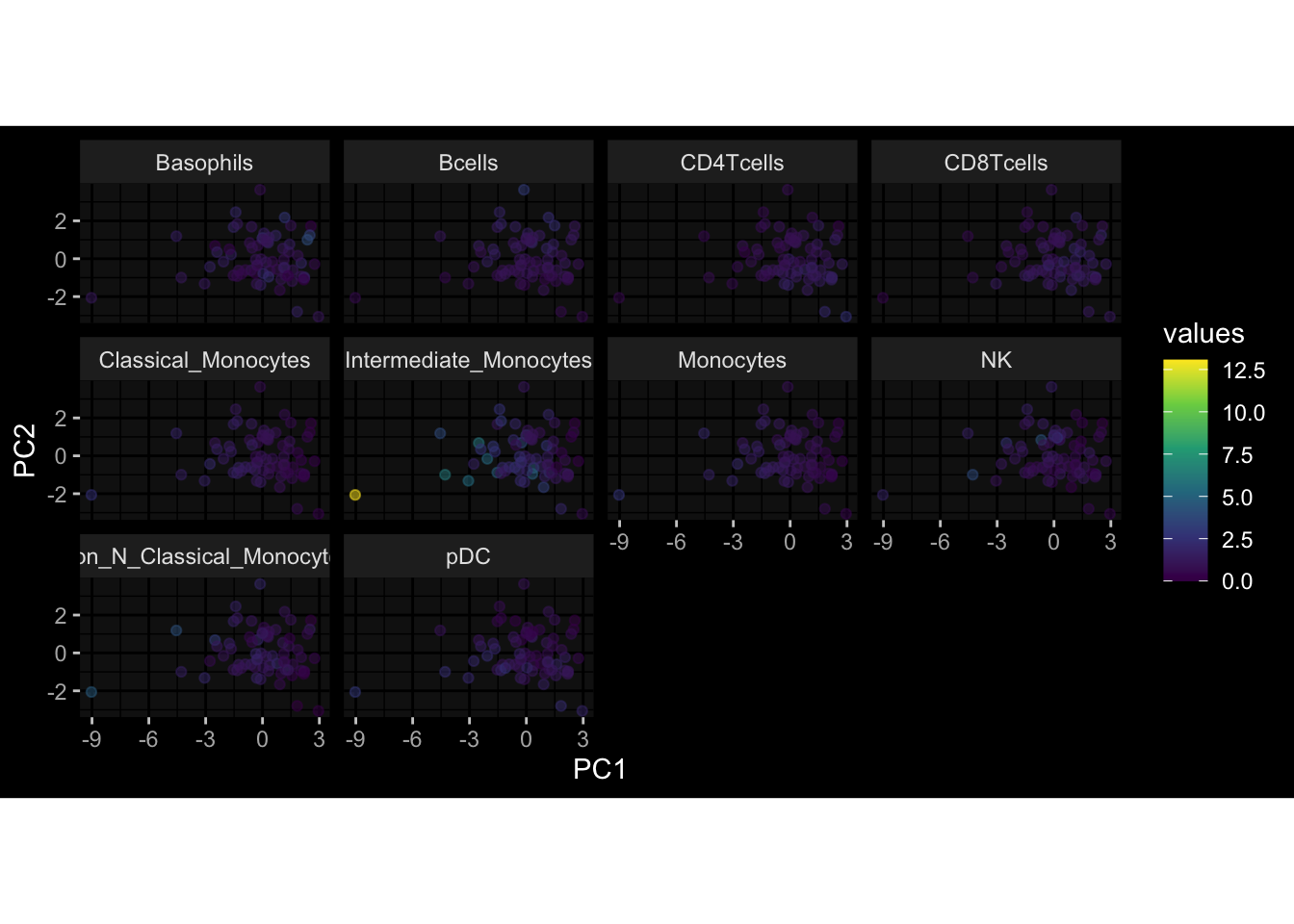
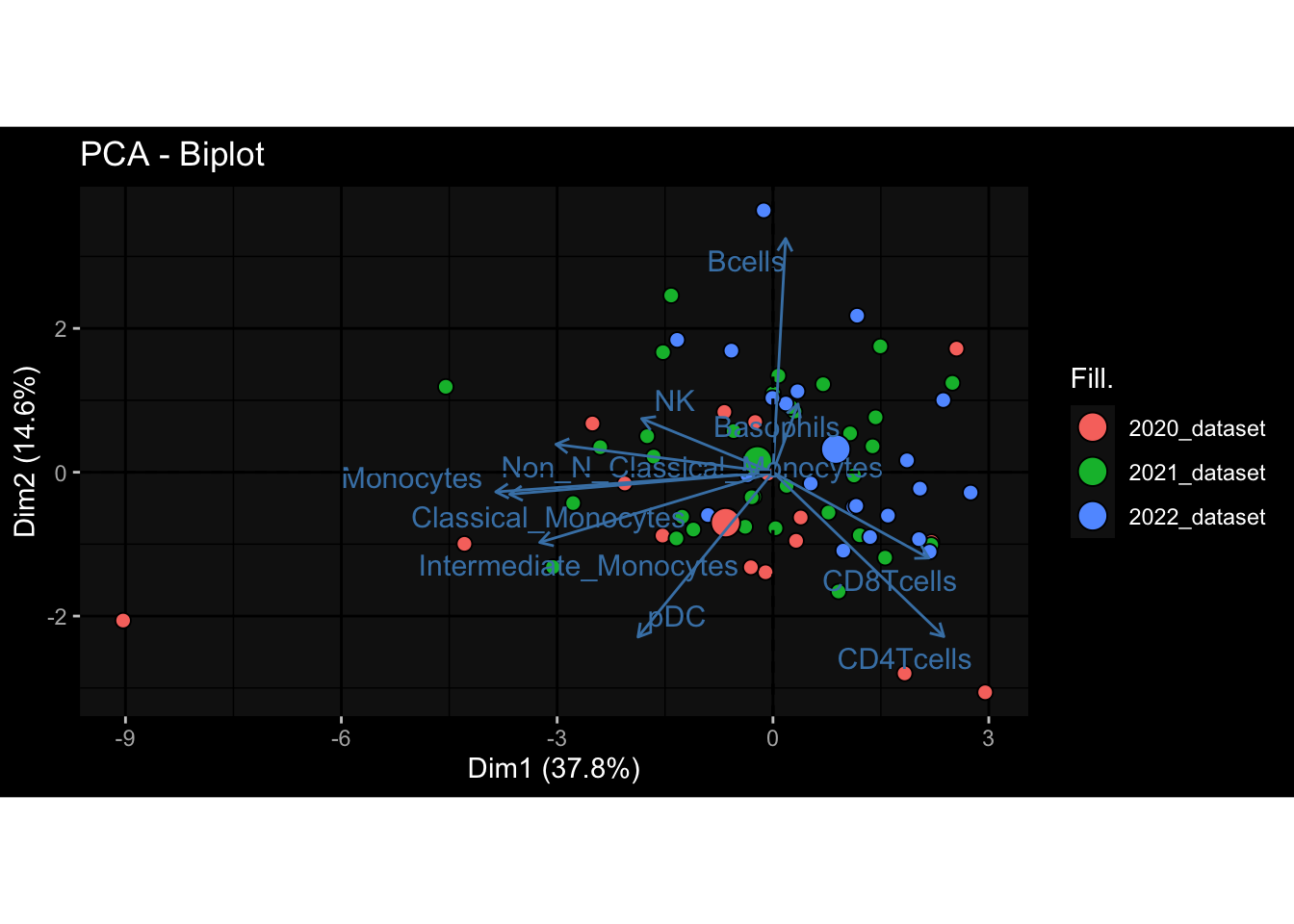
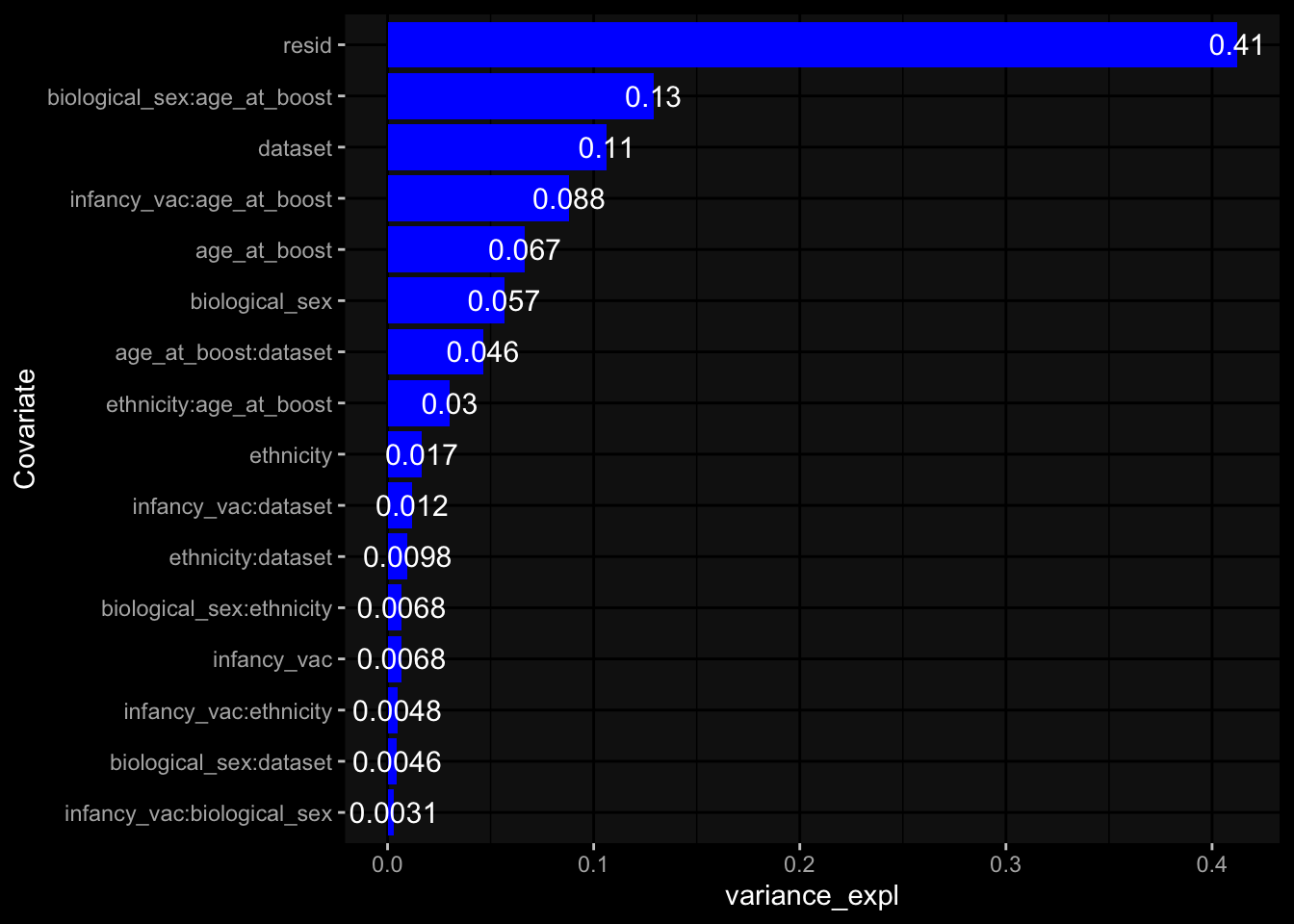
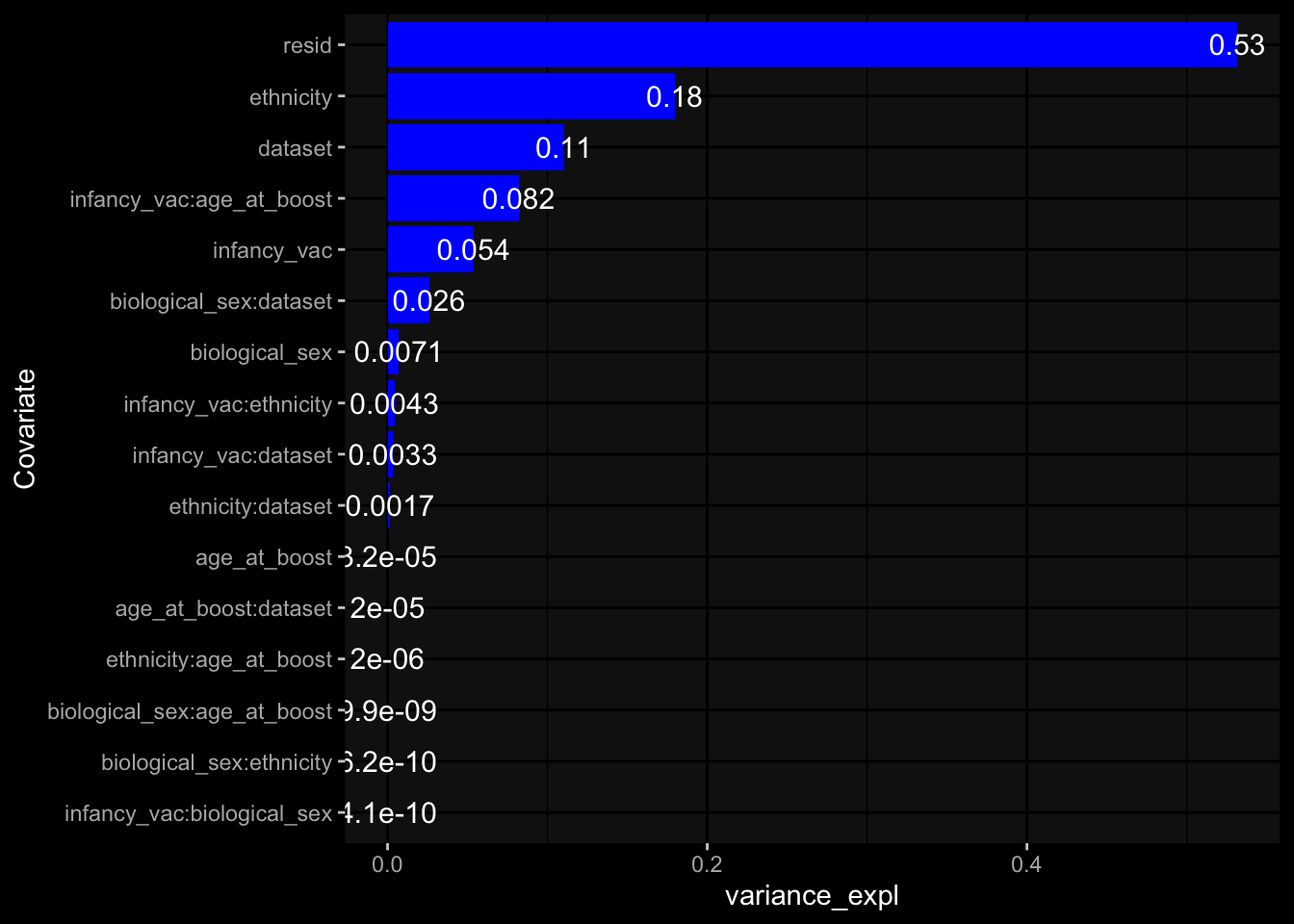
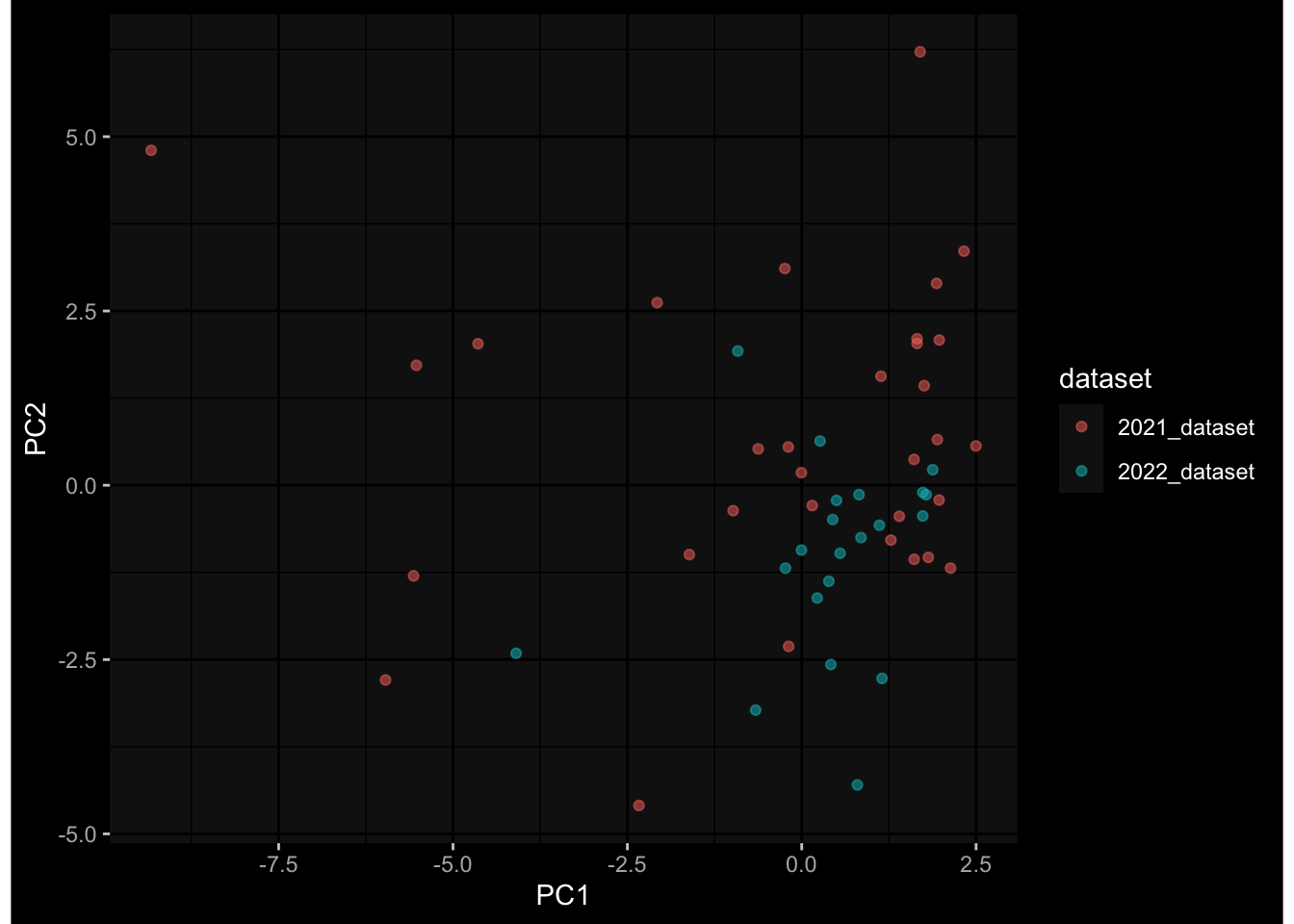
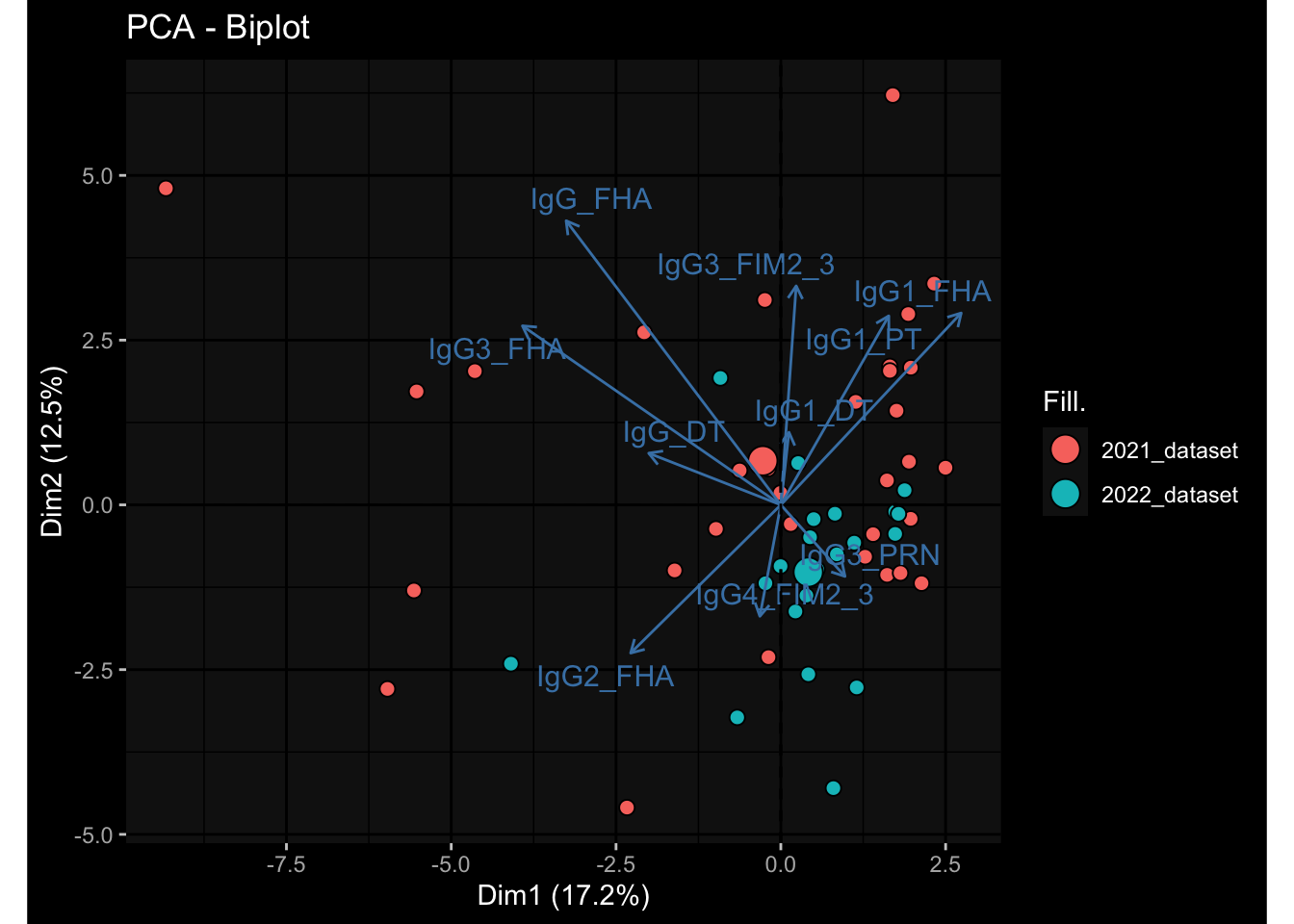
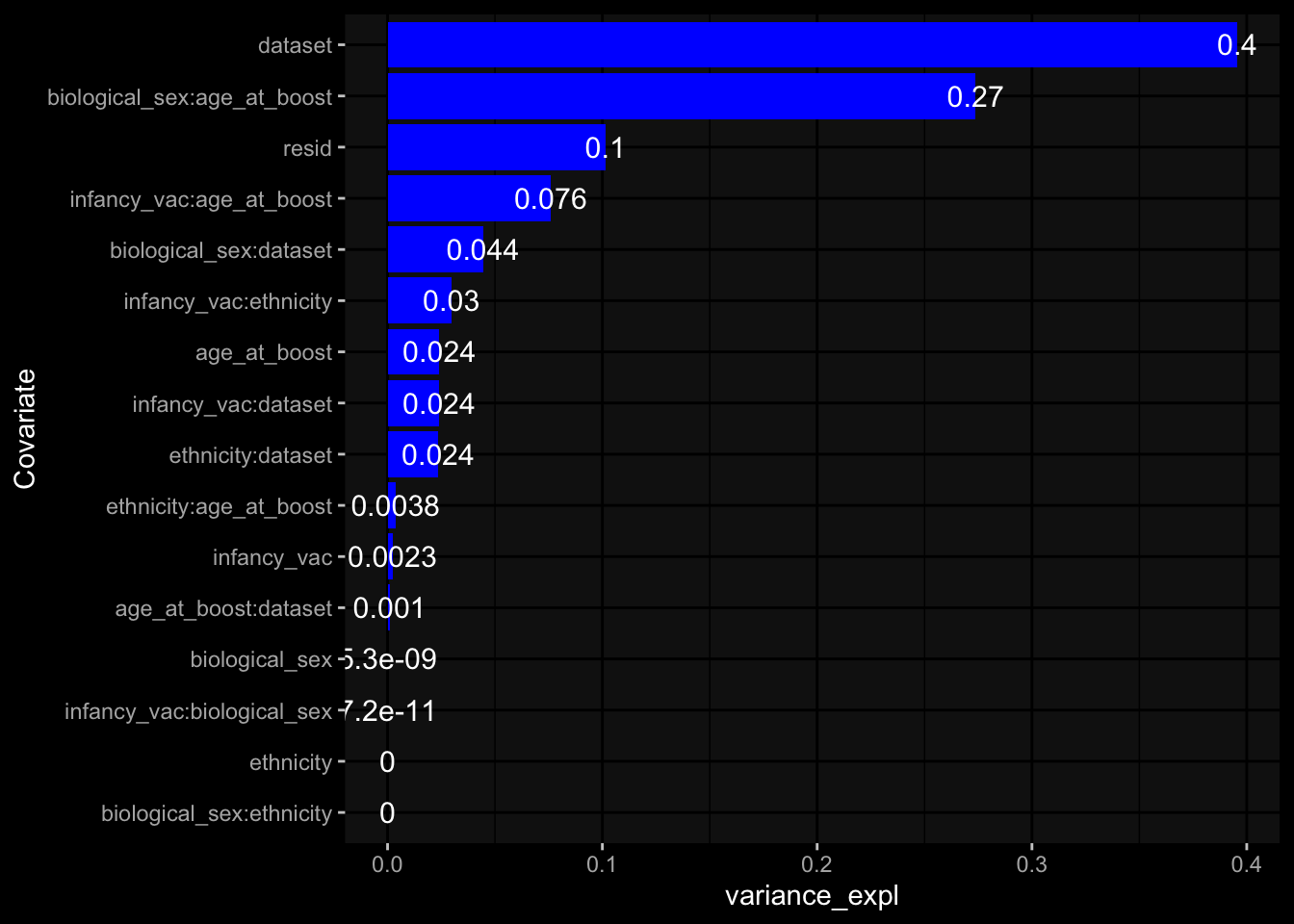
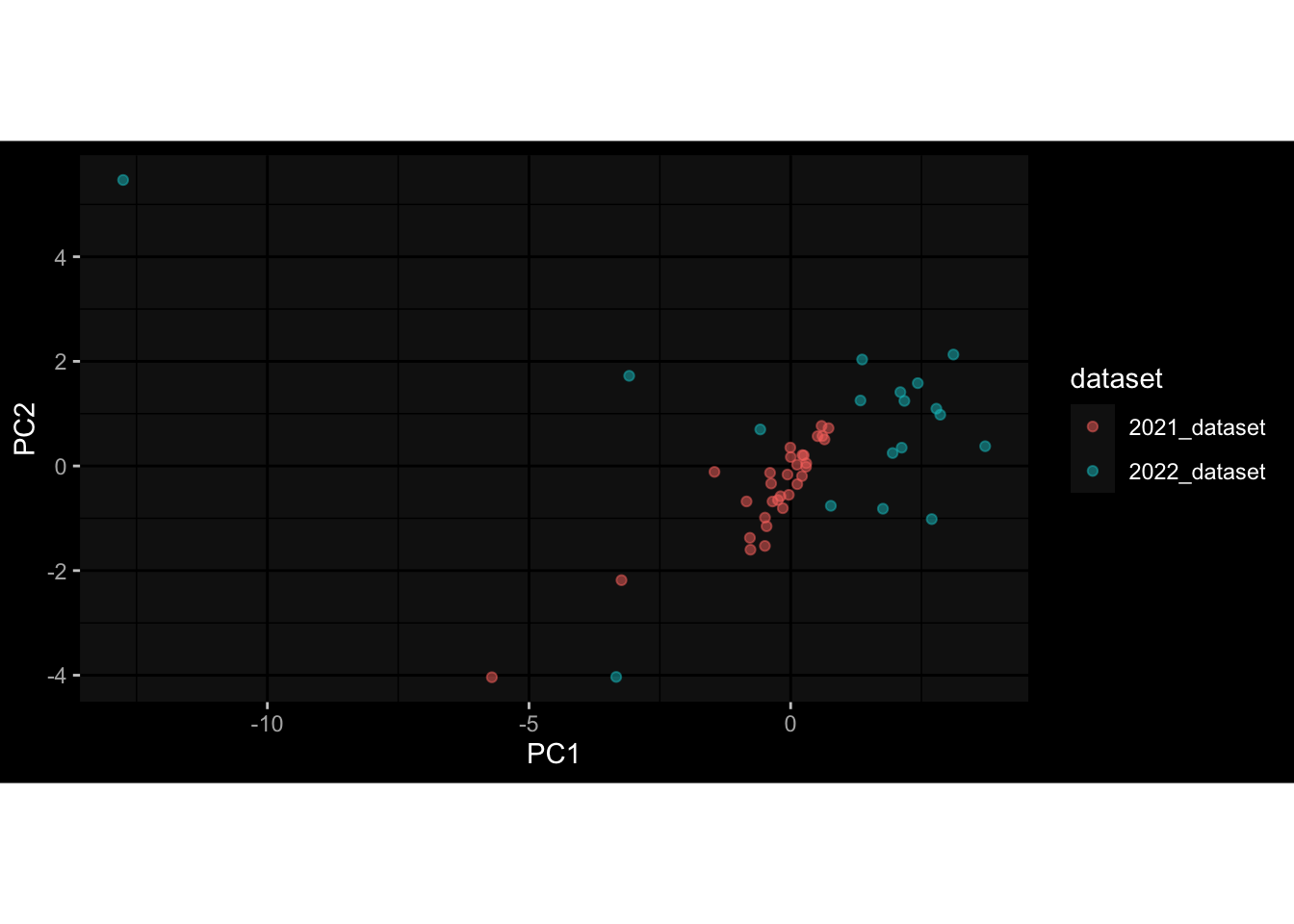
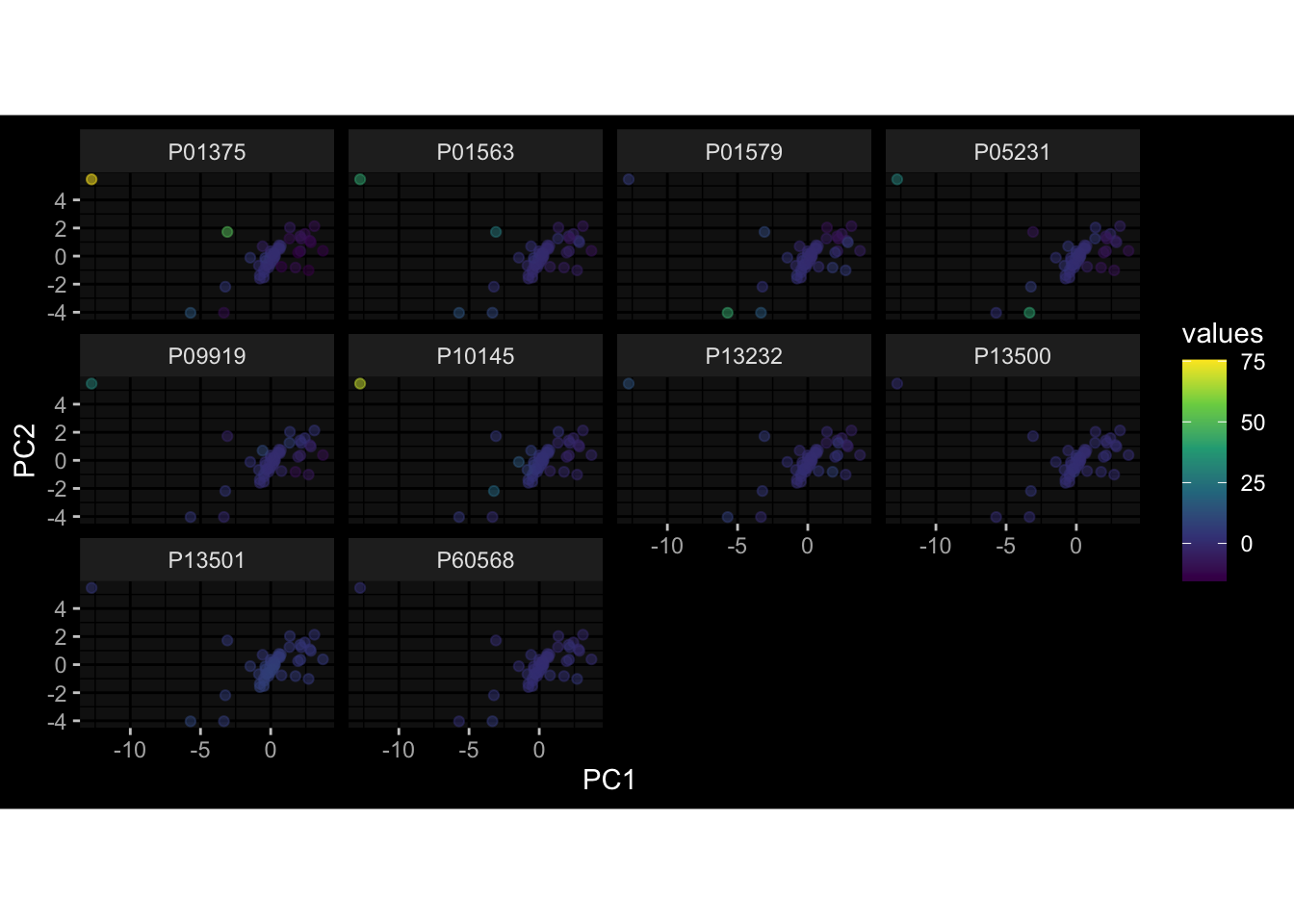
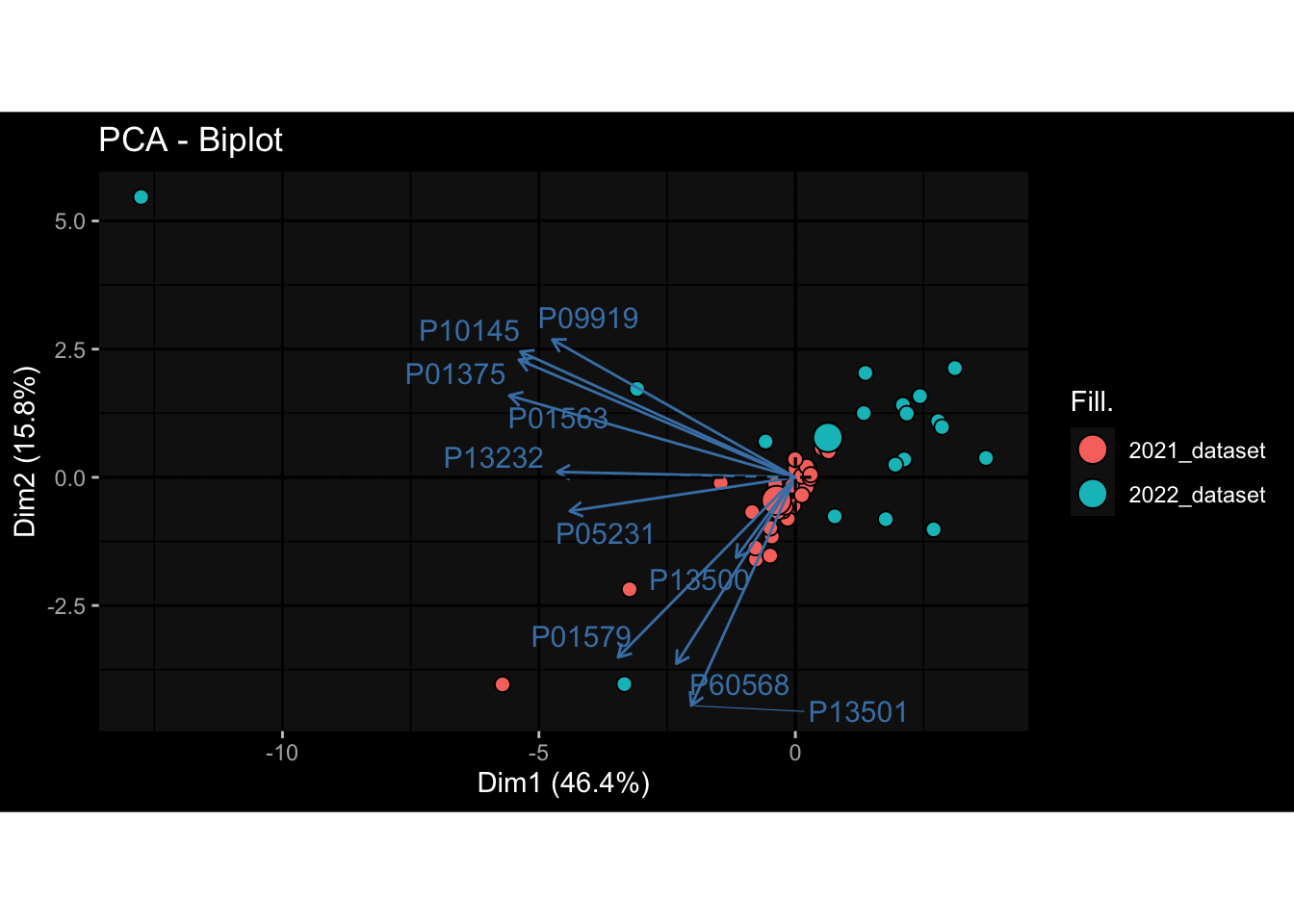
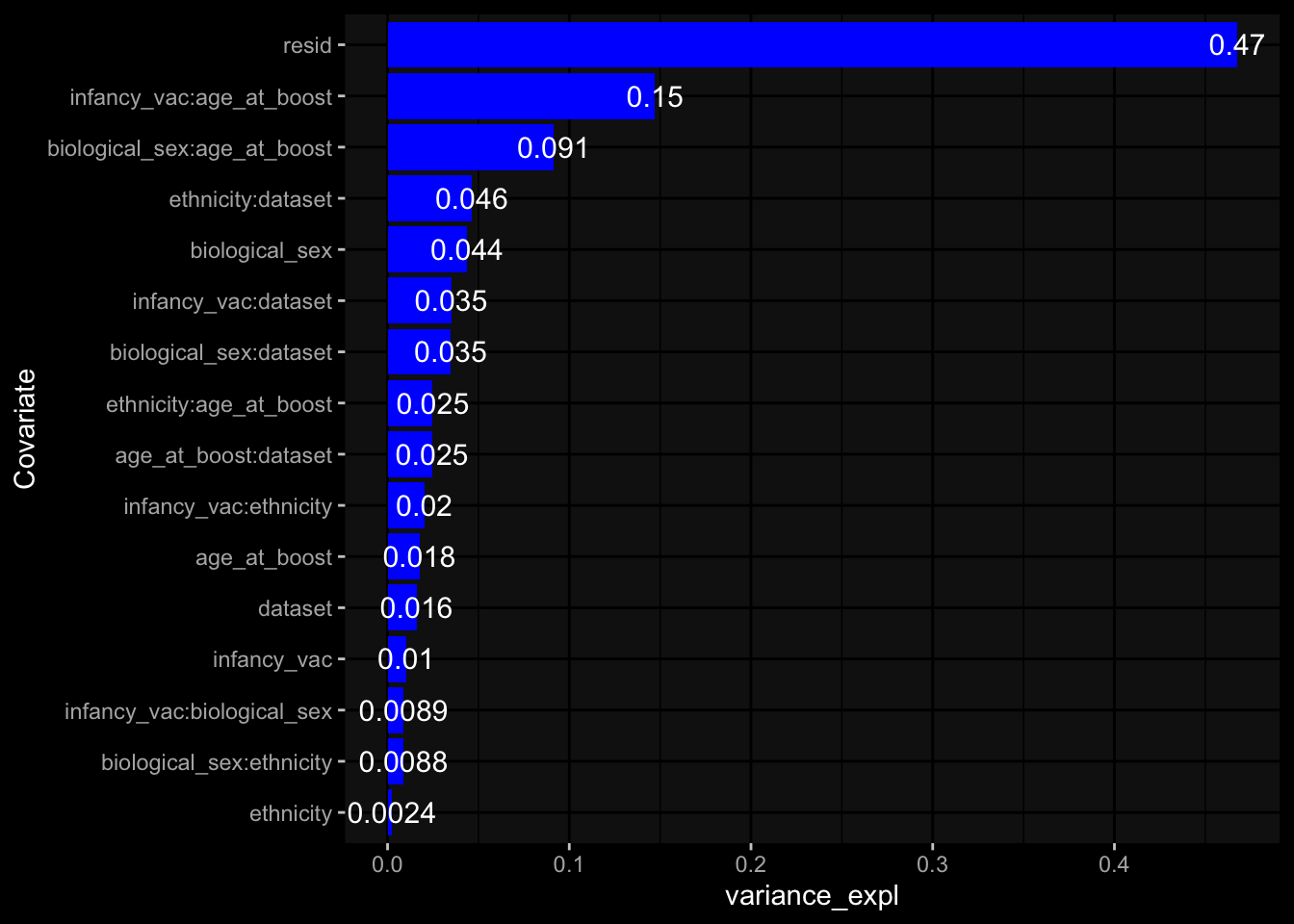
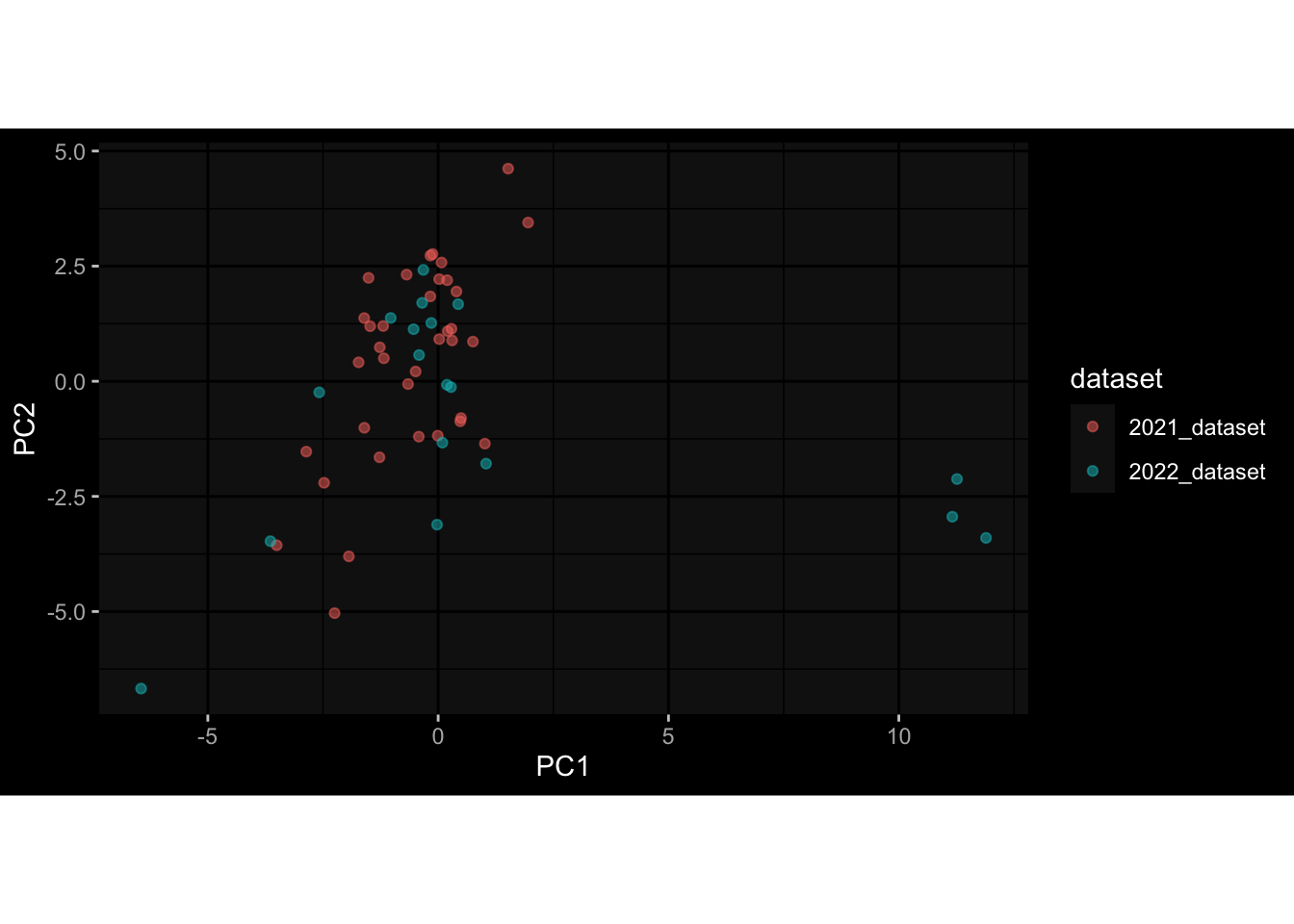
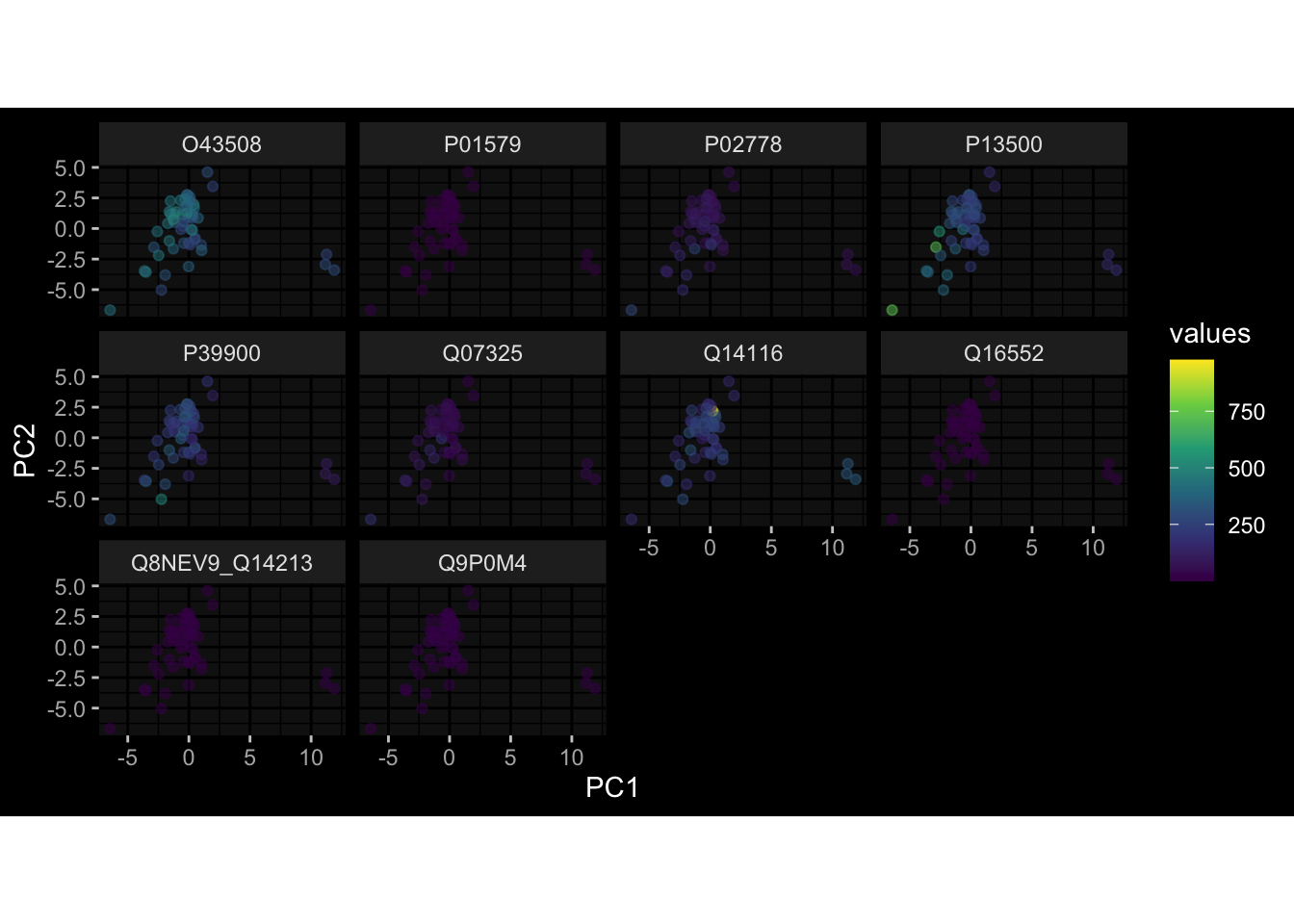
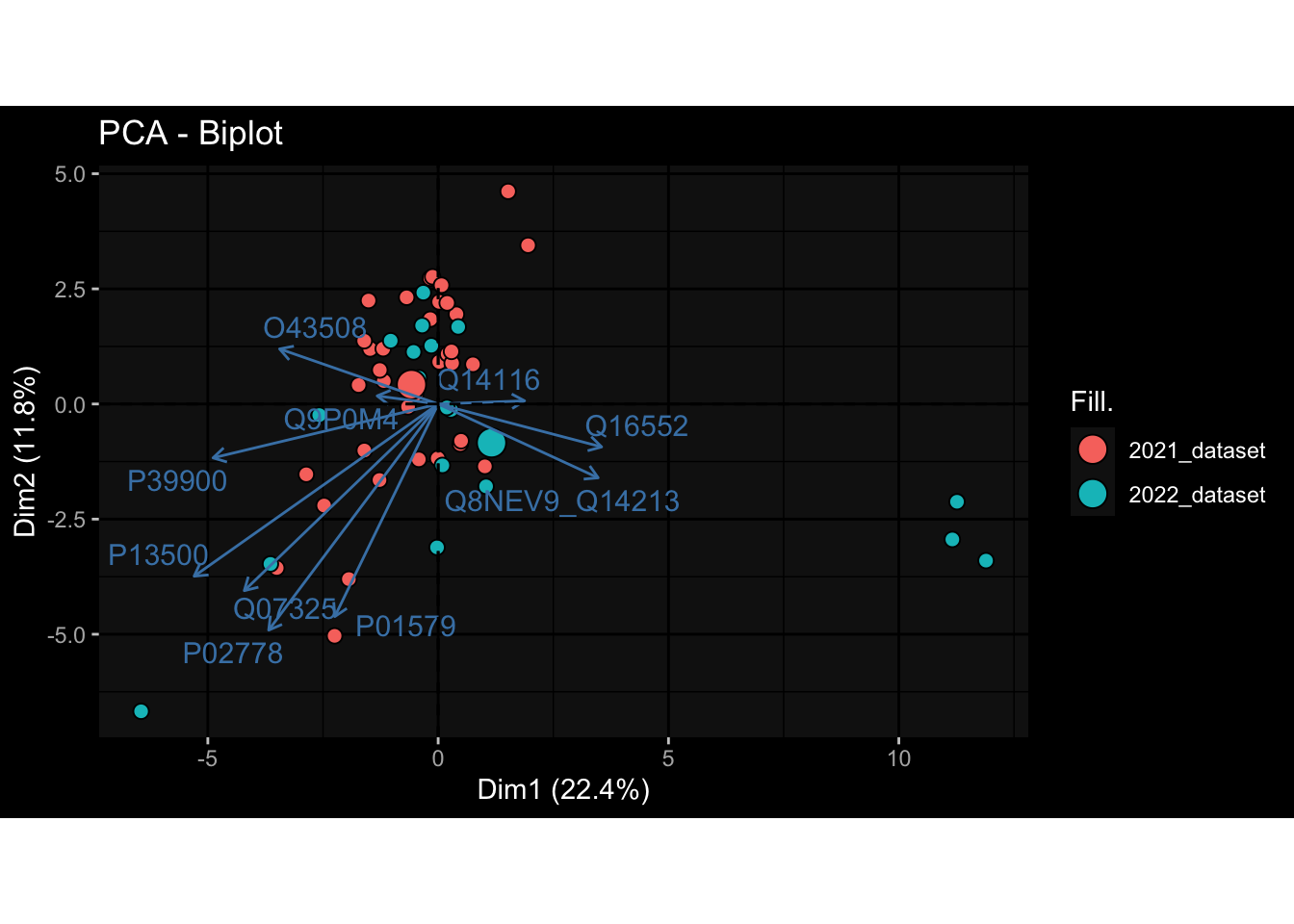
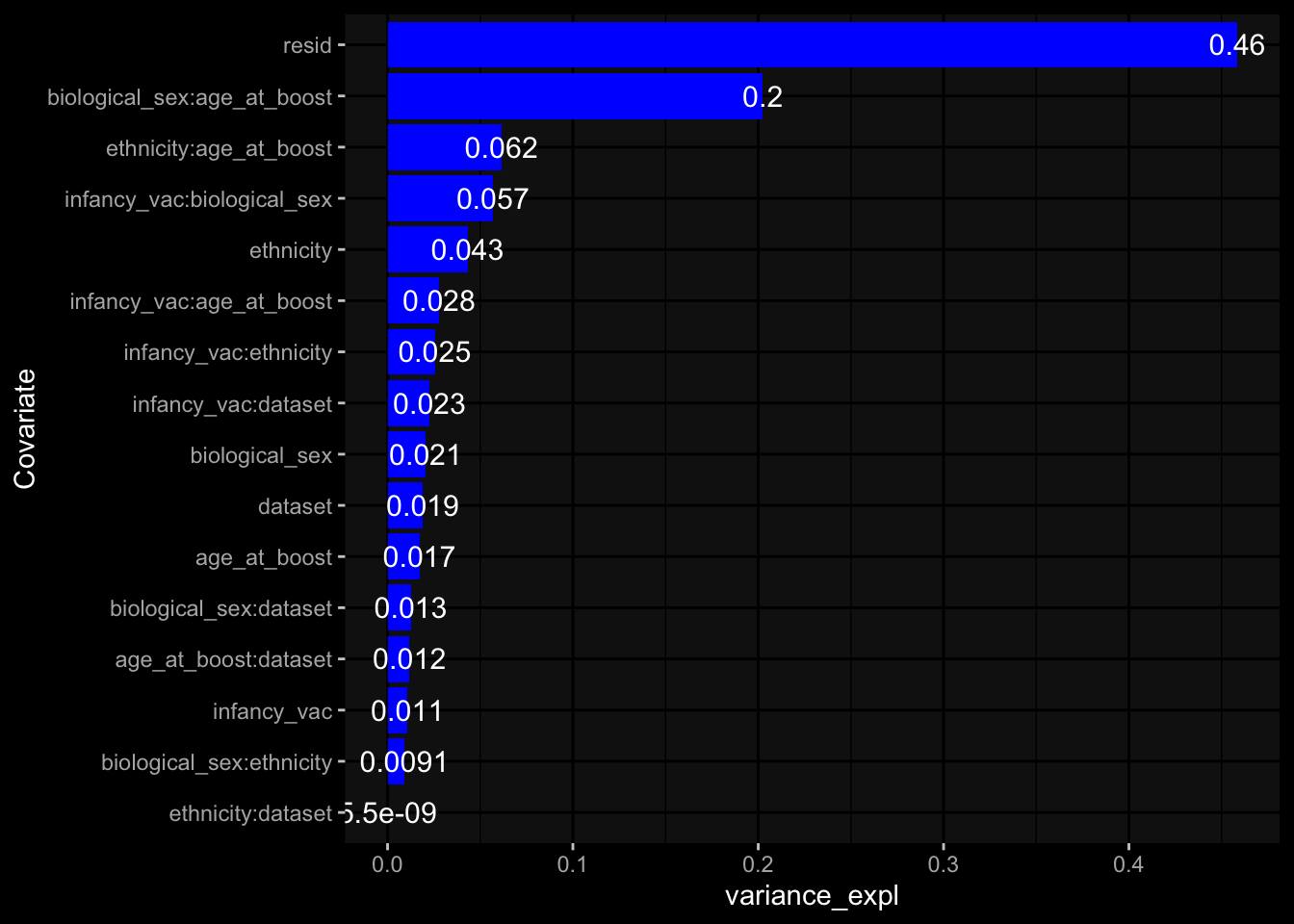
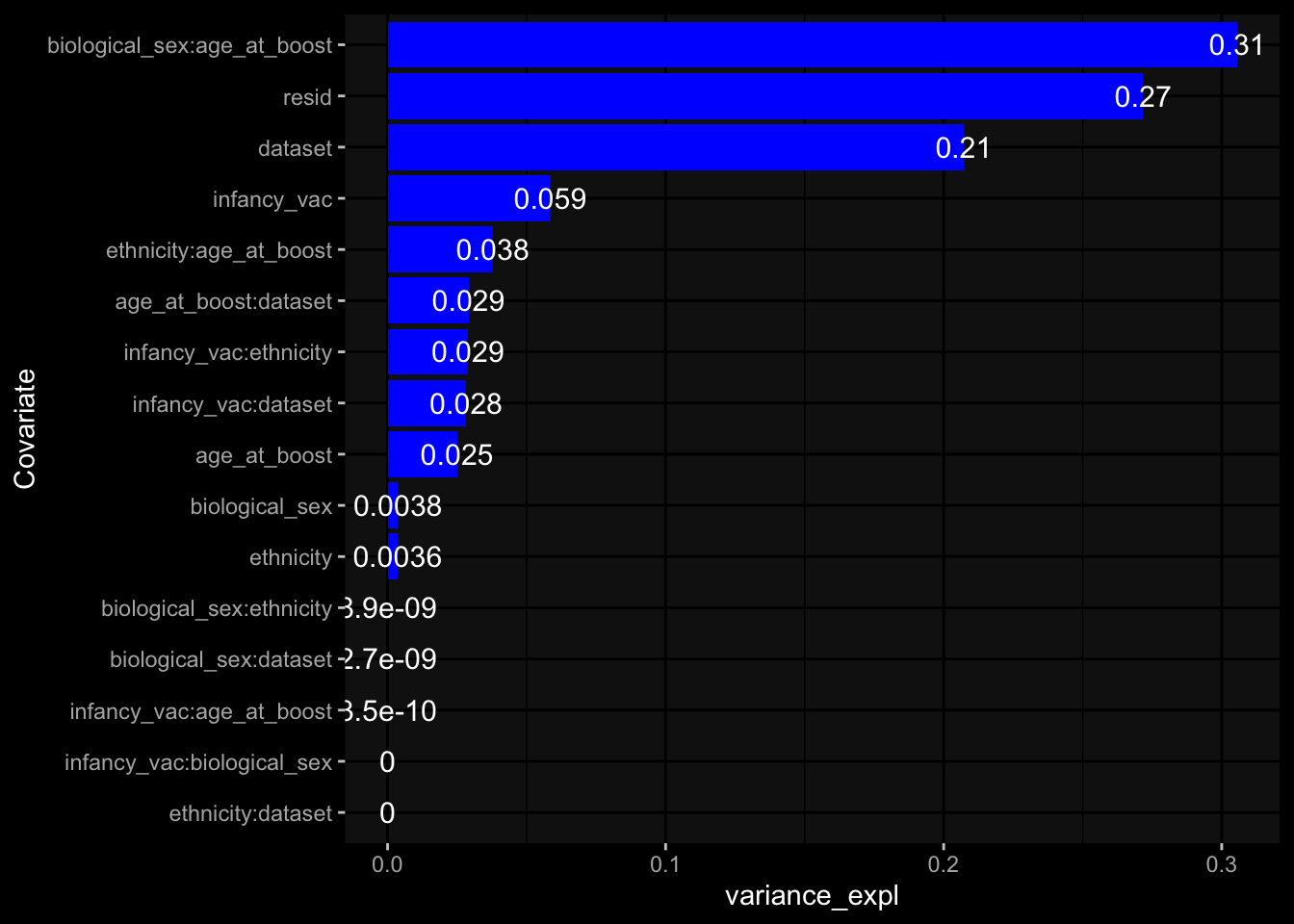
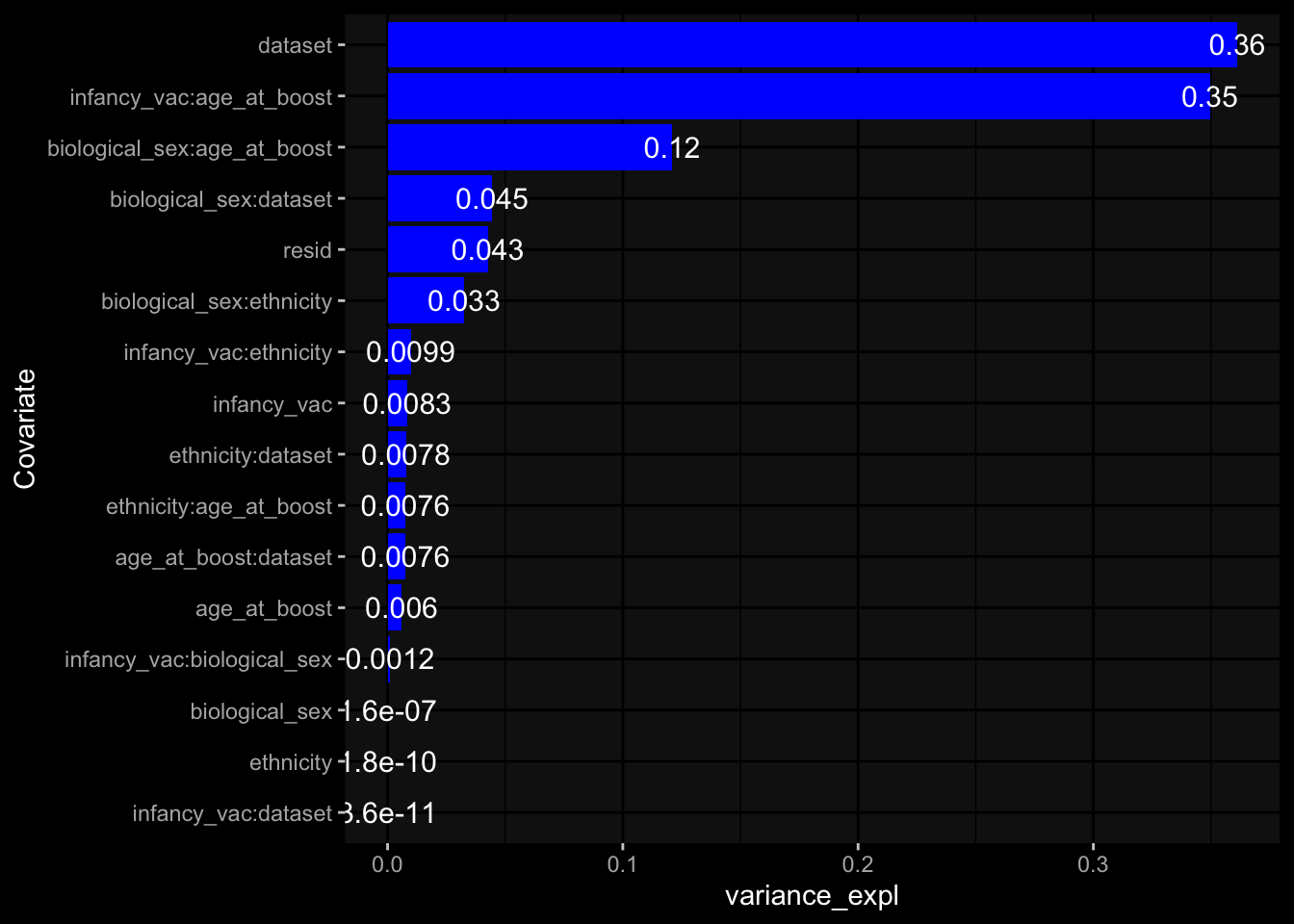
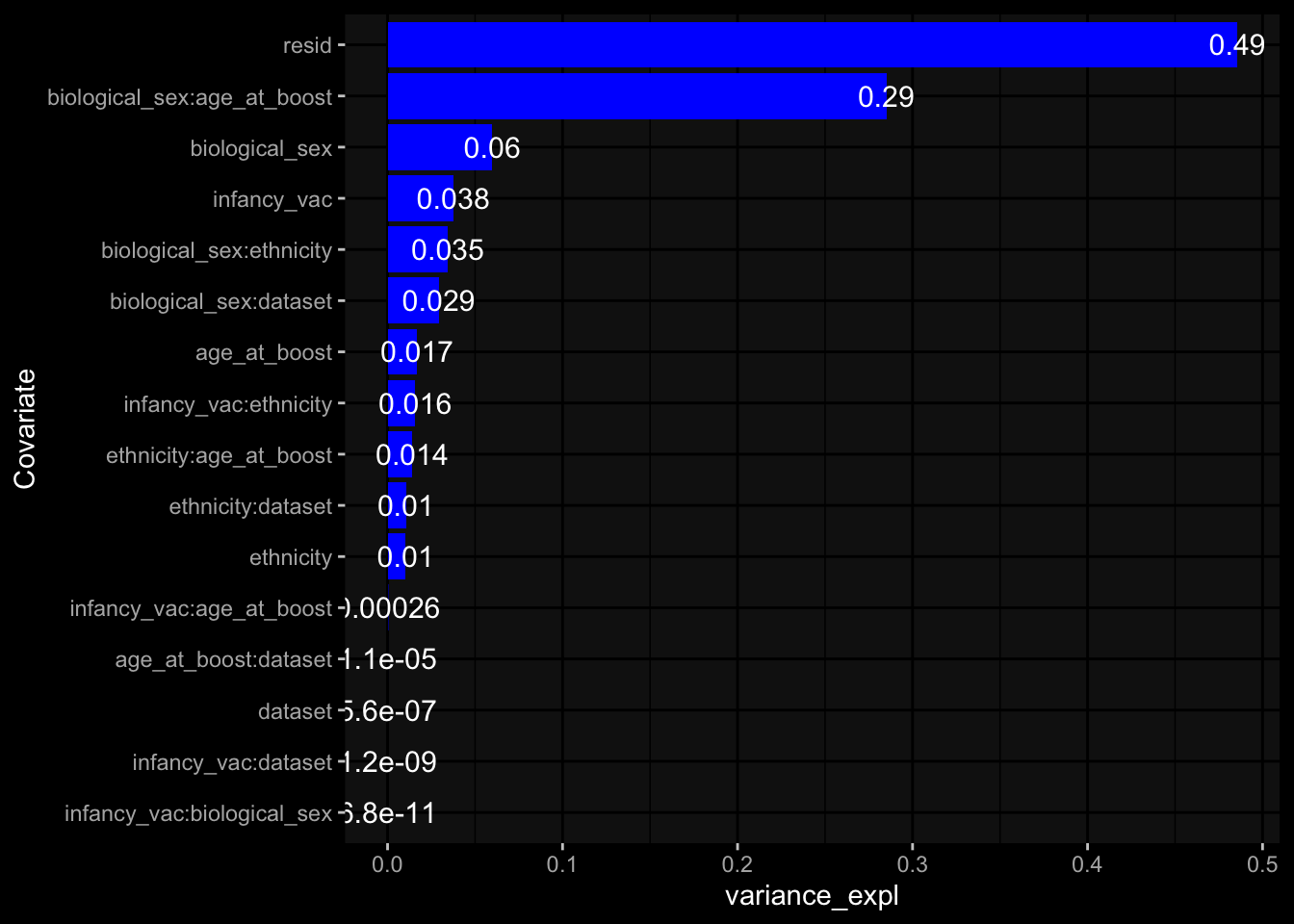
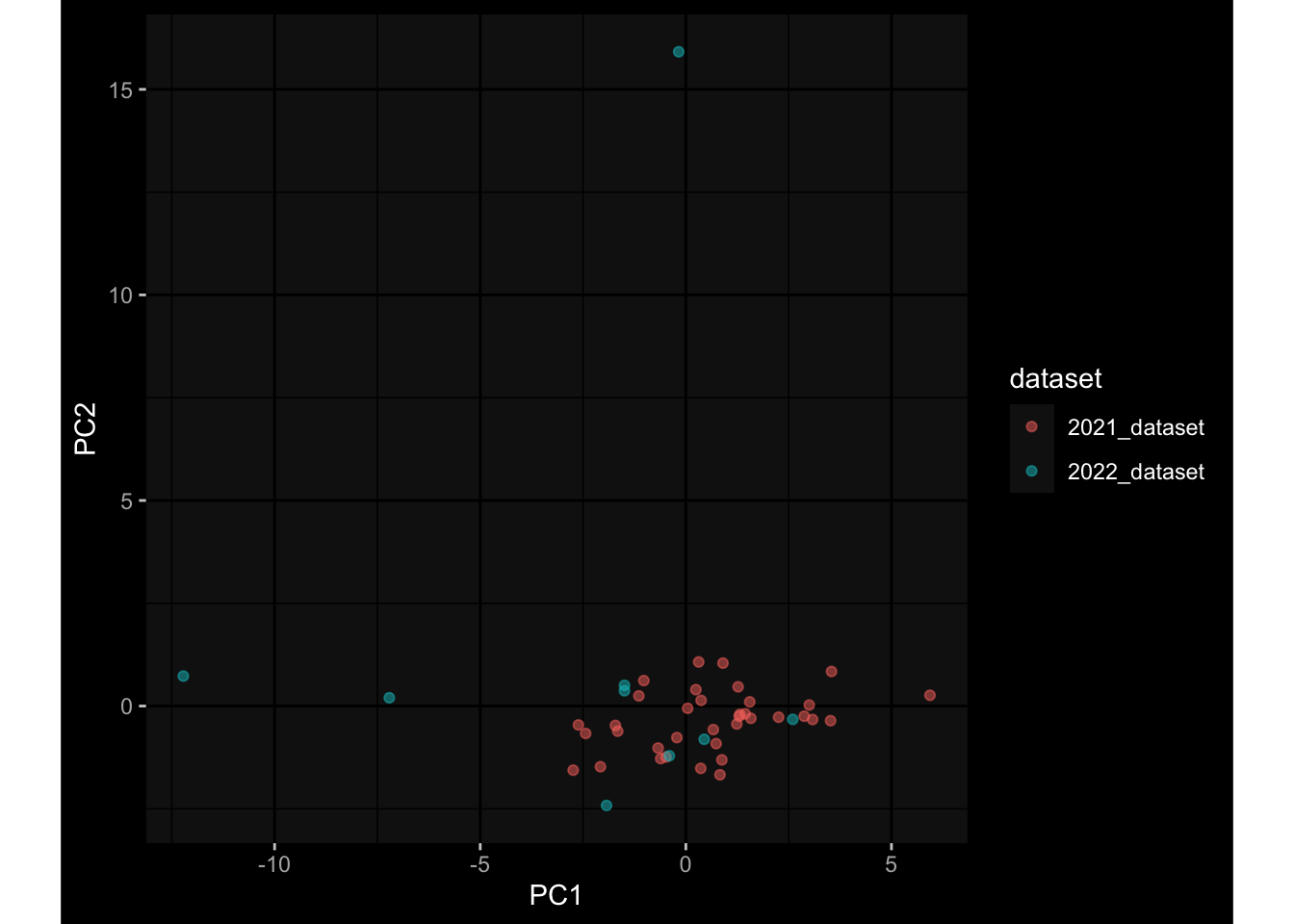
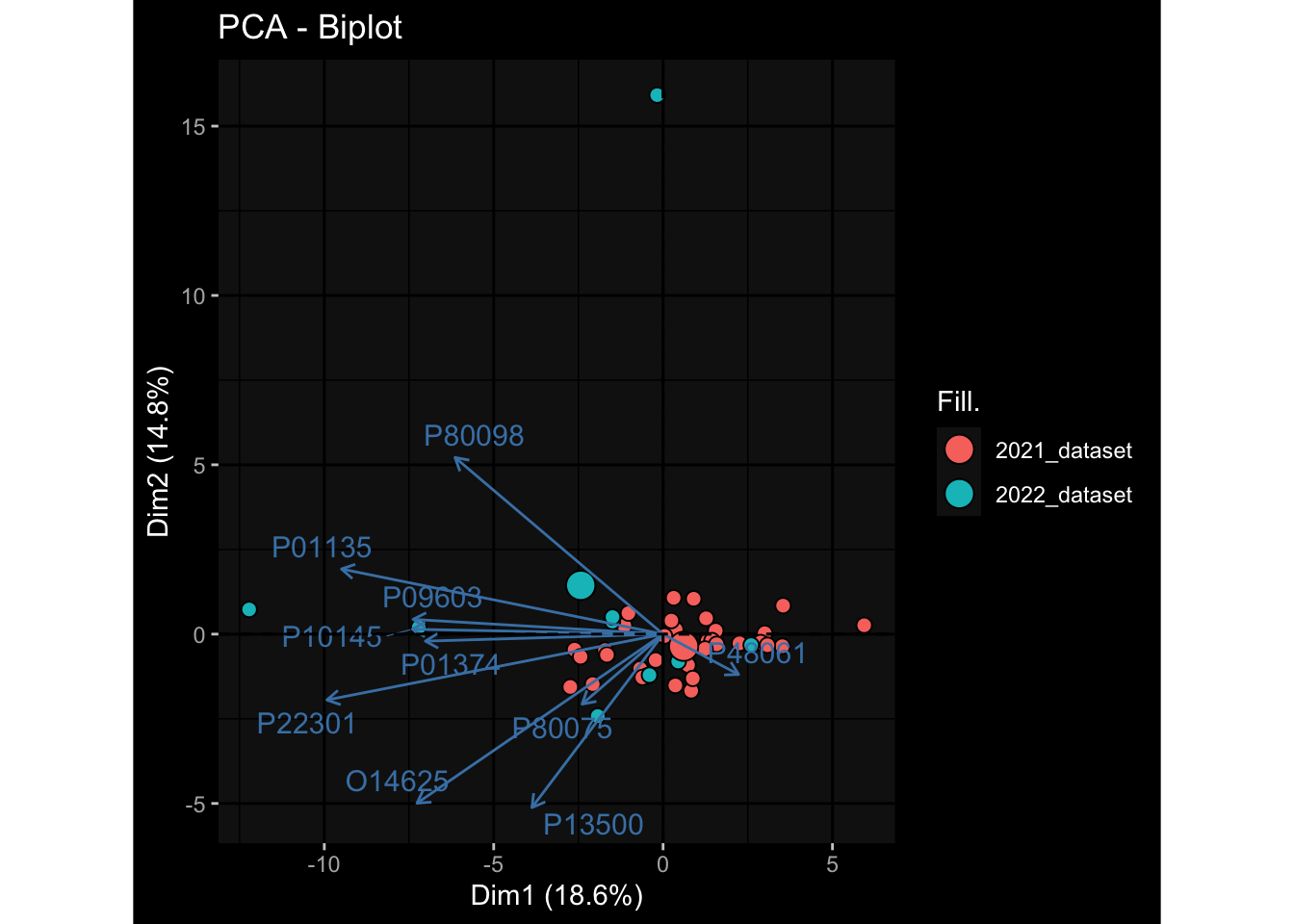
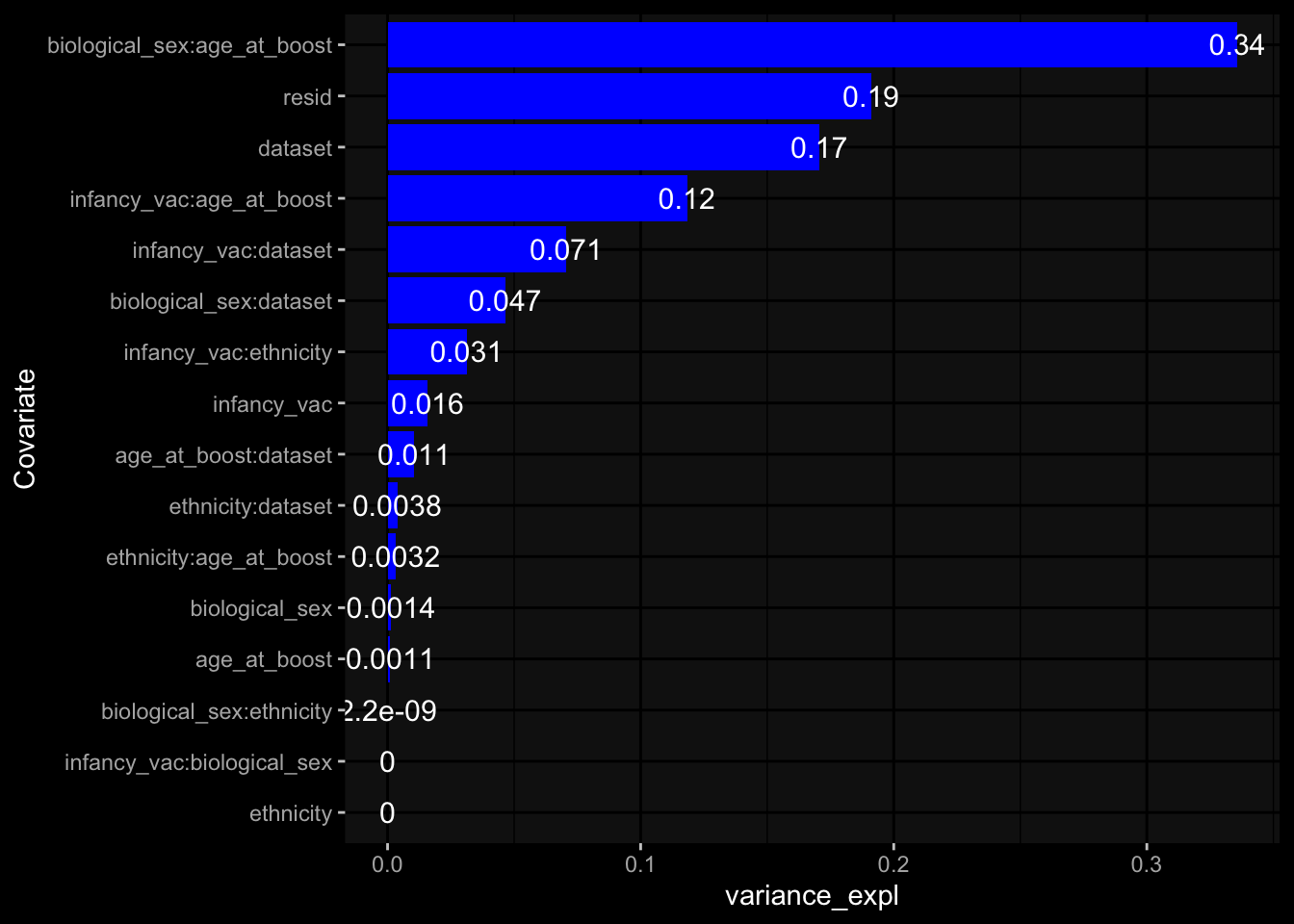
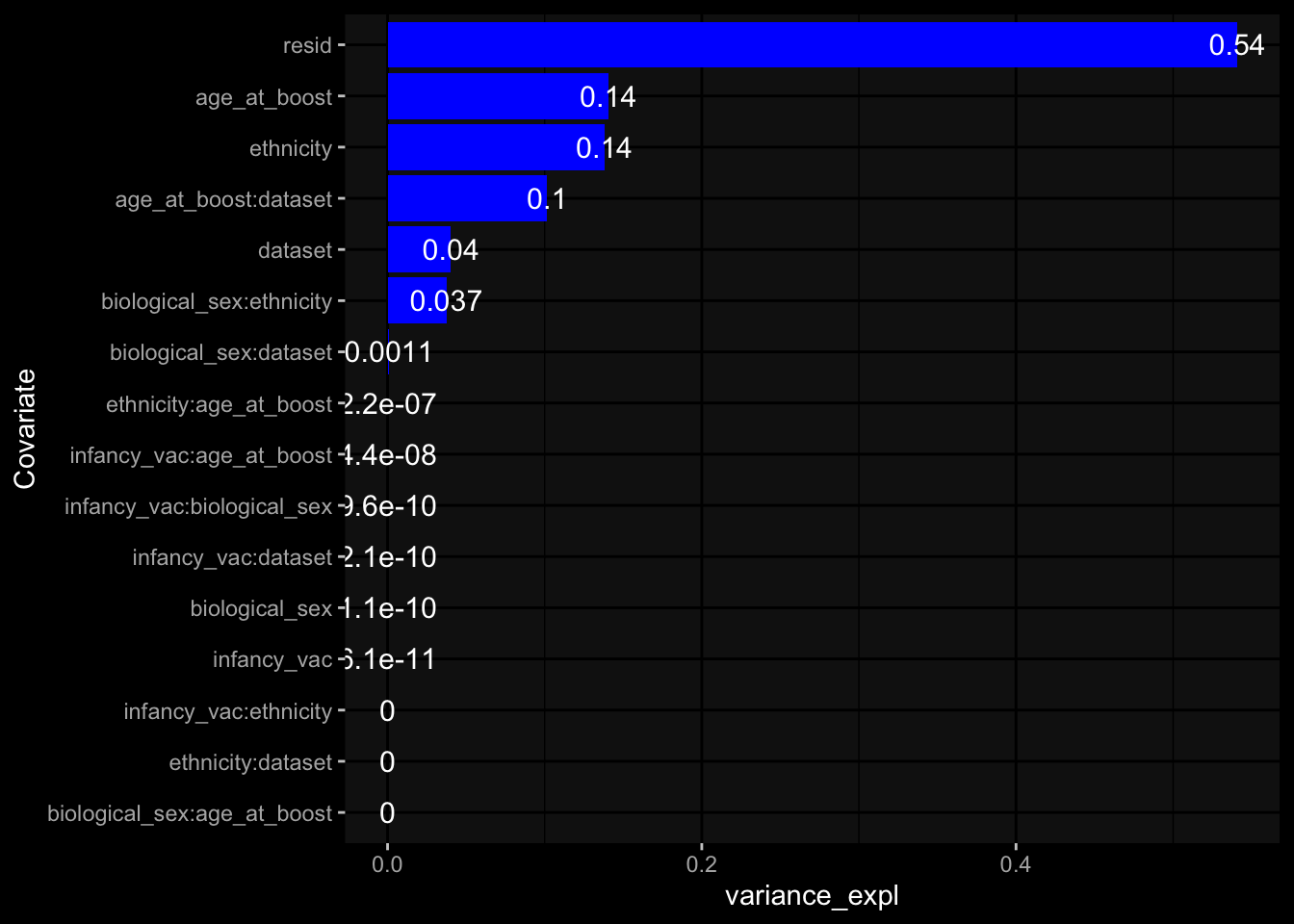
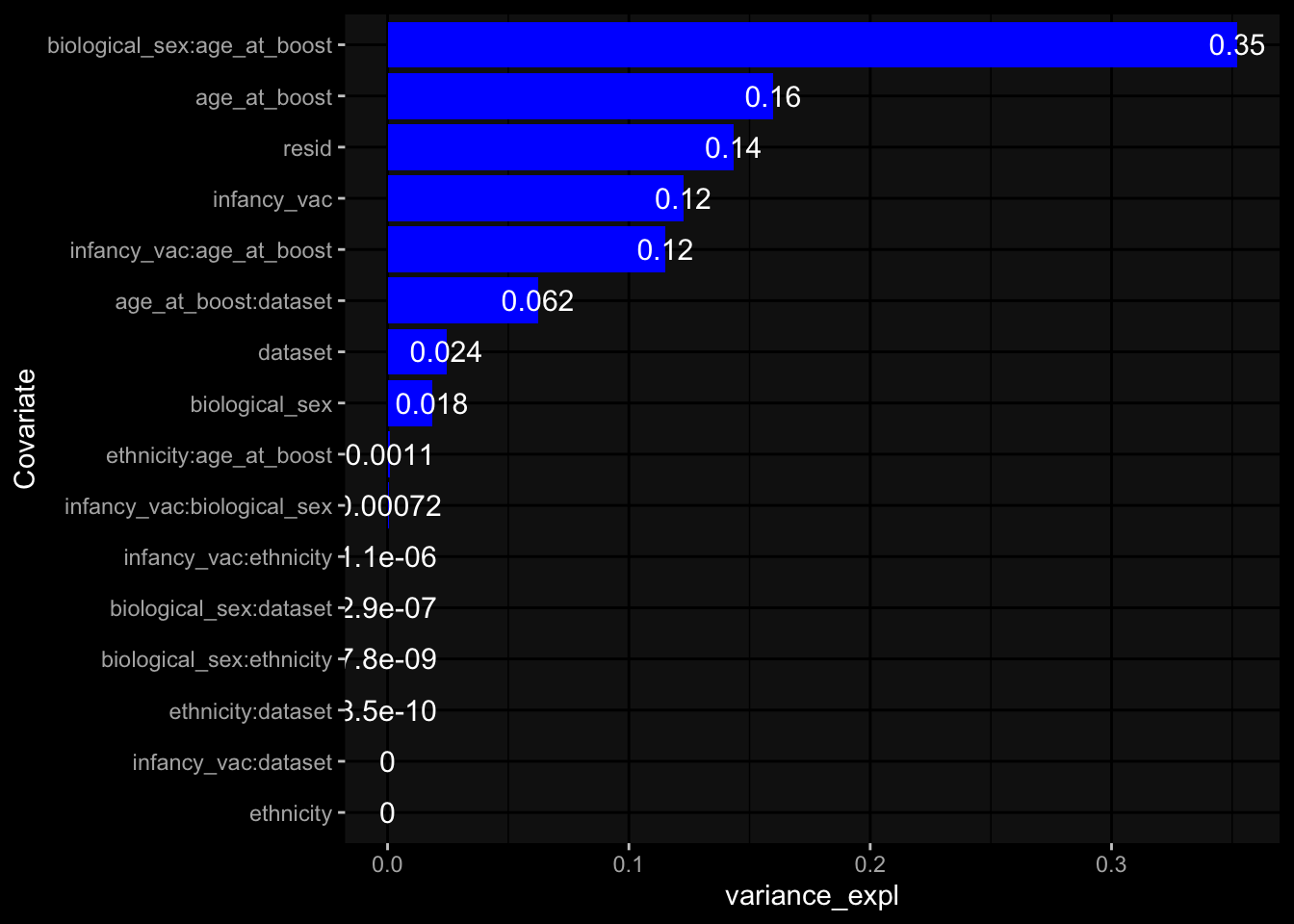
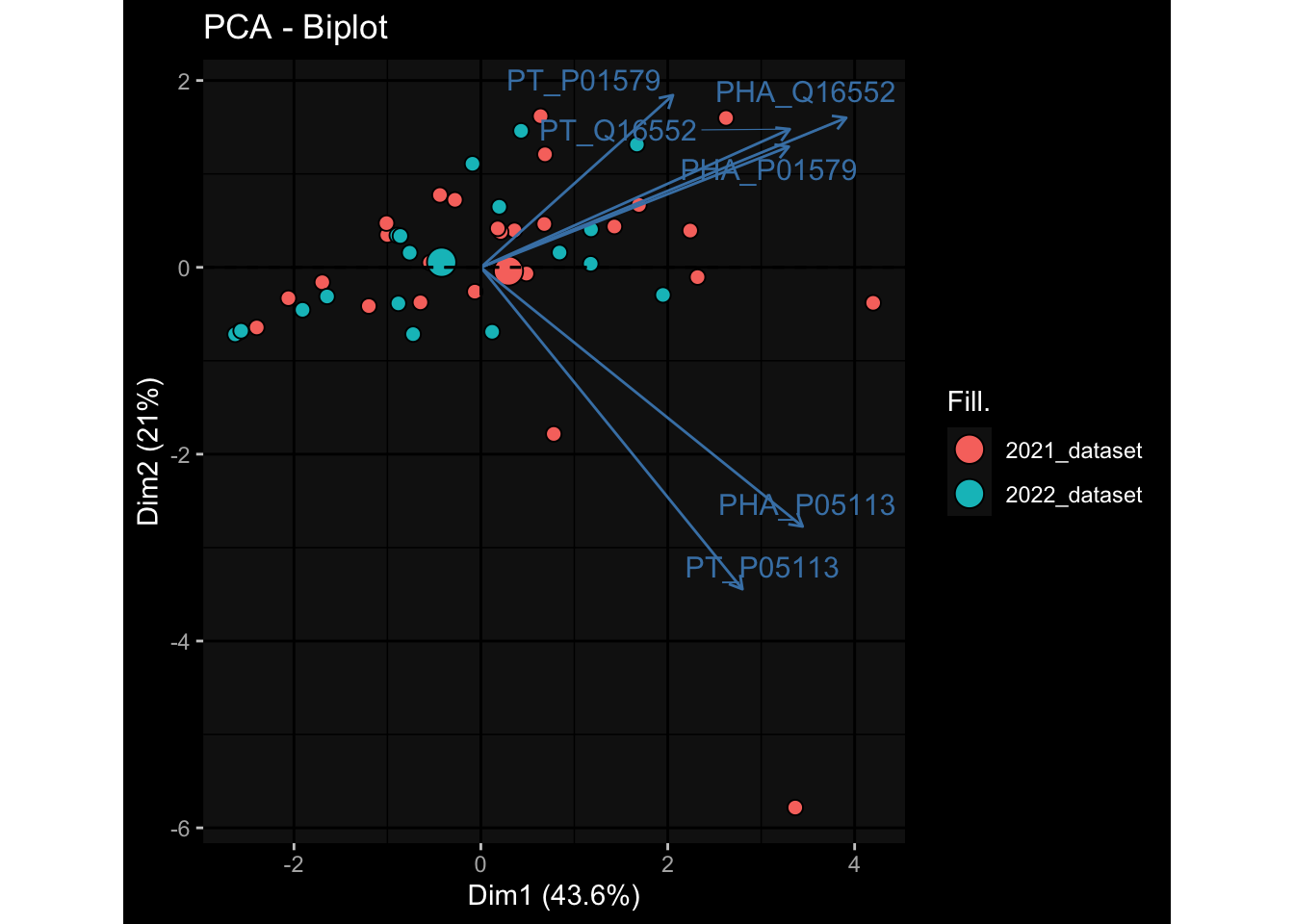
Specimen are only available for baseline, most of the variance is explained by interaction terms age_at_boost:dataset and infancy_vac:dataset. TODO: Think about what to make of this.